
Abstract
Transition metal phosphides hold novel metallic, semimetallic, and semiconducting behaviors. Here we report by ab initio calculations a systematical study on the structural and electronic properties of \(\hbox {MP}_4\) (M = Cr, Mo, W) phosphides in monoclinic C2/c (\(C_{2h}^6\)) symmetry. Their dynamical stabilities have been confirmed by phonon modes calculations. Detailed analysis of the electronic band structures and density of states reveal that \(\hbox {CrP}_4\) is a semiconductor with an indirect band gap of 0.47 eV in association with the p orbital of P atoms, while \(\hbox {MoP}_4\) is a Dirac semimetal with an isolated nodal point at the \(\Gamma\) point and \(\hbox {WP}_4\) is a topological nodal line semimetal with a closed nodal ring inside the first Brillouin zone relative to the d orbital of Mo and W atoms, respectively. Comparison of the phosphides with group VB, VIB and VIIB transition metals shows a trend of change from metallic to semiconducting behavior from \(\hbox {VB-MP}_4\) to VIIB-\(\hbox {MP}_4\) compounds. These results provide a systematical understandings on the distinct electronic properties of these compounds.
Similar content being viewed by others
Introduction
Transition metal phosphides (TMPs) have been attracted considerable research interest due to their structural and compositional diversity that results in a broad range of novel electronic, magnetic and catalytic properties1,2,3,4. This family consists of large number of materials, having distinct crystallographic structures and morphologies because of choices of different TMs and phosphorus atoms5. These compounds have been studied extensively due to their outstanding physical and chemical properties such as high catalytic activity6, good electrical conductivity7, and magnetocaloric behaviors8,9. TMPs have been appeared as an efficient catalyst for hydrogen evolution reduction (HER)4,10,11,12,13. For example, nanowires of FeP and \(\hbox {FeP}_2\) have been used widely for hydrogen evolution in both strong alkaline and acidic aqueous solutions10. CoP11, \(\hbox {CoP}_3\)12, and \(\hbox {MoP}_2\)13 are also reported as an excellent materials for HER and oxygen evolution reduction (OER) due to their good stability. Moreover, phosphorus rich phases have been found more effective for HER and OER, and have better stability because of the presence of a large number of negatively charge P-atom centers14,15. In addition to electrocatalysis process, TMPs have various potential device applications, such as usage in electrotonic components, luminescent and semiconductor devices and as an anode material in lithium-ion batteries16,17,18,19. Moreover, some TMPs such as TaP20 hold topological Weyl semimetal feature, and WP has been recently reported to have Dirac like points near the Fermi level21. Similarly, transition metal diphosphide compounds, like \(\hbox {MoP}_2\) and \(\hbox {WP}_2\), were predicated as type-II Weyl topological semimetals22.
Topological semimetals are not only of fundamental physical interests but also of great potential for future applications in quantum computation and spintronics23,24,25,26,27,28. In topological semimetals, topological non-trivial band crossing points or line (line of nodes) exist in three-dimensional (3D) Brillouin zone (BZ) protected by certain symmetries29,30. It can be classified into Dirac semimetal31, Weyl semimetal32,33 and nodal line semimetal (NLSM)30,34,35,36,37. Driac semimetals have been theoretically predicted and experimentally confirmed in several materials such as \(\hbox {Cd}_3\hbox {As}_2\)31 and \(\hbox {Na}_3\hbox {Bi}\)37. Topological Weyl semimetals have paring two-fold degenerate Weyl points with opposite distinct chiralities that may be right handed or left handed and have been realized in the materials breaking the time reversal (T) symmetry such as pyrochlore iridate33 or spatial inversion (P) symmetry such as TaAs family of compounds38. In NLSMs, the bands crossing points form continuous line rather than discrete points, generally enforced due to the band inversion mechanism39,40 and protected by PT symmetry34. Topological NLSMs have been found in \(\hbox {CaP}_3\)41, \(\hbox {Ca}_3\hbox {P}_2\)42 phosphides and 3D graphene network structures43,44,45,46,47,48,49,50,51,52,53,54, etc.
In this paper, based on ab initio calculations, we systematically investigate the transition metal phosphides \(\hbox {MP}_4\) (\(\hbox {M} = \hbox {Cr}\), Mo, W) for the structural stability and electronic properties. These three compounds are all in monoclinic phase with C2/c (\(C_{2h}^6\)) symmetry, while \(\hbox {CrP}_4\) and \(\hbox {MoP}_4\) have been experimentally synthesized55 and \(\hbox {WP}_4\) is not yet reported. Their mechanical stabilities are confirmed with phonon mode analysis. Electronic band calculations show that \(\hbox {CrP}_4\) is a semiconductor with an indirect band gap of 0.47 eV, \(\hbox {MoP}_4\) is a topological Dirac semimetal with isolated band crossing at the \(\Gamma\) point, and \(\hbox {WP}_4\) is a topological nodal line semimetal with a closed nodal ring inside the first BZ. We also make a comparison of the phosphides with group VB and VIIB transition metals and a trend of change from metallic to semiconducting is observed from \(\hbox {VB-MP}_4\) to VIIB-\(\hbox {MP}_4\) compounds.
Results and discussion
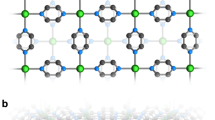
We first present the structural characterization. Figure 1a shows the structure of monoclinic compounds of \(\hbox {MP}_4\) (\(\hbox {M} = \hbox {Cr}\), Mo, W) in C2/c (\(C_{2h}^6\), No. 15) symmetry. The M atoms are depicted in black occupying the 4e Wyckoff positions, while there are two kinds of P atoms (\(\hbox {P}_1\) and \(\hbox {P}_2\)) depicted in orange occupying two distinct 8f Wyckoff positions, respectively, as listed in Table 1. The metals environments in \(\hbox {MP}_4\) compounds can be described as the octahedral coordination environment, in which metal atoms are always octahedrally surrounded by P atoms, while P atoms have tetrahedrally coordinated environment. Basically, the crystalline structure of monoclinic \(\hbox {MP}_4\) compounds can be seen as a layered structure of black phosphorus in which metal atoms are inserted56 between two buckled phosphorus layers (Fig. 1b). Metal atoms intercalate and reorder the atomic stacks similar to Na atom insertion in black phosphorus57. A sandwiched structure is formed where wave like metal atom layers are in between the two buckled phosphorus layers.

Crystal structure of \(\hbox {MP}_4\) (M = Cr, Mo, W) compounds. (a) The unit cell in monoclinic C2/c symmetry. (b) the layered view. The M atoms are depicted in black while the P atoms are depicted in orange. These structures were drawn using VESTA package76.
There are three unique types of bonds in monoclinic compounds \(\hbox {MP}_4\), namely M-\(\hbox {P}_1\), M-\(\hbox {P}_2\), and \(\hbox {P}_1\)-\(\hbox {P}_2\) chemical bonds. In \(\hbox {CrP}_4\), the bond lengths are 2.277–2.373 Å for Cr-\(\hbox {P}_1\), 2.316 Å for Cr-\(\hbox {P}_2\), and 2.215–2.240 Å for \(\hbox {P}_1\)-\(\hbox {P}_2\); in \(\hbox {MoP}_4\), the bond lengths are 2.396–2.456 Å for Mo-\(\hbox {P}_1\), 2.456 Å for Mo-\(\hbox {P}_2\), and 2.208–2.243 Å for \(\hbox {P}_1\)-\(\hbox {P}_2\); while in \(\hbox {WP}_4\), the bond lengths are 2.398–2.477 Å for W-\(\hbox {P}_1\), 2.453 Å for W-\(\hbox {P}_2\), and 2.215–2.245 Å for \(\hbox {P}_1\)-\(\hbox {P}_2\). Meanwhile, there are three distinct types of bond angles depicted as \(\angle \hbox {P}_1\)-M-\(\hbox {P}_1\), \(\angle \hbox {P}_2\)-M-\(\hbox {P}_2\) and \(\angle \hbox {P}_1\)-M-\(\hbox {P}_2\). For \(\hbox {CrP}_4\), the bond angles are \(90.03^{\circ }\) for \(\angle \hbox {P}_1\)-Cr-\(\hbox {P}_1\), \(85.30^{\circ }\) for \(\angle \hbox {P}_2\)-Cr-\(\hbox {P}_2\), and \(92.37^{\circ }\) for \(\angle \hbox {P}_1\)-Cr-\(\hbox {P}_2\); for \(\hbox {MoP}_4\), the bond angles are \(88.19^{\circ }\) for \(\angle \hbox {P}_1\)-Mo-\(\hbox {P}_1\), \(83.80^{\circ }\) for \(\angle \hbox {P}_2\)-Mo-\(\hbox {P}_2\), and \(94.0^{\circ }\) for \(\angle \hbox {P}_1\)-Mo-\(\hbox {P}_2\); while for \(\hbox {WP}_4\), the bond angles are \(88.19^{\circ }\) for \(\angle \hbox {P}_1\)-W-\(\hbox {P}_1\), \(84.16^{\circ }\) for \(\angle \hbox {P}_2\)-W-\(\hbox {P}_2\), and \(93.83^{\circ }\) for \(\angle \hbox {P}_1\)-W-\(\hbox {P}_2\). It can be seen that the bond lengths between P-P atoms are almost same in the three \(\hbox {MP}_4\) compounds, while the bond lengths between Mo-P and W-P atoms are clearly larger than that between Cr-P atoms. Meanwhile, \(\angle \hbox {P}_1\)-M-\(\hbox {P}_2\) are found larger than the other angles in all \(\hbox {MP}_4\) compounds. The calculated equilibrium lattice parameters, bond lengths, and bond angles for \(\hbox {MP}_4\) compounds are listed in Table 2. It is seen that our calculated structural parameters matches well with the reported experimental and calculated data55,58,59.
To examine the dynamical stability of \(\hbox {MP}_4\) compounds, we have calculated the phonon band structures and partial phonon density of states (PDOS) with equilibrium lattice parameters in a \(2\times 2\times 2\) supercell, as shown in Fig. 2. For \(\hbox {CrP}_4\), \(\hbox {MoP}_4\) and \(\hbox {WP}_4\), no imaginary frequencies occur in the whole BZ and PDOS, thus confirming the structural stability of the three compounds. There are some similarities in the phonon band structures and PDOS for \(\hbox {CrP}_4\), \(\hbox {MoP}_4\) and \(\hbox {WP}_4\) due to the same space symmetry groups and elementary components for the three compounds. The highest vibrational frequencies all happen near the \(\Gamma\) point and the values are 519.8 cm\(^{-1}\) for \(\hbox {CrP}_4\), 521.8 cm\(^{-1}\) for \(\hbox {MoP}_4\) and 526.8 cm\(^{-1}\) for \(\hbox {WP}_4\), respectively. It is seen from the PDOS that the lower frequency modes are mainly contributed by the metal atoms because of their heavier masses while the higher frequency modes are mainly contributed by the P atoms with lighter masses.

Phonon band structures and density of states (PDOS) for \(\hbox {MP}_4\) (M = Cr, Mo, W) compounds at equilibrium lattice parameters. The lower frequency modes are mainly contributed by the metal atoms because of their heavier masses while the higher frequency modes are mainly contributed by the P atoms with lighter masses.

Electronic band structures for (a) \(\hbox {CrP}_4\), (b) \(\hbox {MoP}_4\) and (c) \(\hbox {WP}_4\) at equilibrium lattice parameters using HSE06 functional (without spin-orbital coupling). (d) The BZ with several high-symmetry points indicated at \(\Gamma\) (0.00, 0.00, 0.00), Y (0.3067, 0.3067, 0.0440), F (0.3631, 0.3631, 0.3937), H (0.2503, 0.2503, 0.6943), Z (0.00, 0.00, 0.50), I (0.50, \(-0.50\), 0.50), and X (0.50, \(-0.50\), 0.00). The nodal ring (green circle) in (d) is formed by band crossing points for \(\hbox {WP}_4\) compound were plotted using MATLAB software.
Next we discuss the electronic properties of \(\hbox {MP}_4\) (\(\hbox {M} = \hbox {Cr}\), Mo, W) compounds. Figure 3 represents the calculated electronic band structures along the high symmetry directions of the BZ using HSE06 functional60 and the fermi energy (\(E_F\)) is set to zero. For \(\hbox {CrP}_4\) as shown in Fig. 3a, the conduction band minimum (CBM) is located along H-Z direction and valence band maximum (VBM) is located along F-H direction, showing a semiconducting behavior with an indirect band gap of 0.47 eV, which is smaller than the reported direct band gap of 0.63 eV58. For \(\hbox {MoP}_4\) as shown in Fig. 3b, the lowest conduction band and highest valence band are degenerate at \(\Gamma\) point near the \(E_F\), indicating that \(\hbox {MoP}_4\) is a Dirac semimetal with a four-fold degenerate Dirac point at the \(\Gamma\) point61. Moreover, our calculations show that the valence and conduction bands of \(\hbox {WP}_4\) exhibit linear dispersion near the \(E_F\) and cross along the \(\Gamma\)-X high symmetry direction (Fig. 3c) due to the band inversion mechanism39,40. To further explore the topological electronic properties, we establish a tight binding (TB) model using the maximally localized Wannier functions (MLWFs)62,63 to search the nodal points in the 3D BZ. We find that the nodal points (or band crossing points) of valence and conduction bands in \(\hbox {WP}_4\) form a continuous nodal ring in the full BZ (see Fig. 3d), thus, \(\hbox {WP}_4\) can be termed as a topological nodal line semimetal with a closed nodal ring protected by PT symmetry34,35,41.
It is interesting to notice that although Cr, Mo and W are all in the VIB group of the Periodic Table of Elements, \(\hbox {CrP}_4\) is an indirect band gap semiconductor, \(\hbox {MoP}_4\) is a Dirac semimetal with a single nodal point, and \(\hbox {WP}_4\) is a nodal line semimetal with a closed nodal ring. The metallicity of \(\hbox {CrP}_4\), \(\hbox {MoP}_4\), and \(\hbox {WP}_4\) grows with the increase of the elementary ordinal from 3d to 5d transition metals. To further understand the electronic properties, we have plotted the total and partial density of states (DOS) of \(\hbox {MP}_4\) compounds as shown in Fig. 4. For \(\hbox {CrP}_4\) (Fig. 4a), there is a band gap of 0.47 eV as depicted in Fig. 3a. The states around the Fermi level are mainly contributed by the p states of P atoms (Fig. 4b), relative to the covalent bonds between P-P atoms. For \(\hbox {MoP}_4\) (Fig. 4c), there is a little peak on the Fermi level, the states at the Fermi level are mainly composed of d orbital of Mo atoms (see Fig. 4d). Moreover, for \(\hbox {WP}_4\) (Fig. 4e), there is a little peak on the Fermi level, but larger than that in \(\hbox {MoP}_4\), the states at the Fermi level are predominantly composed of P-p orbital and W-d orbital (Fig. 4f). It can be inferred that the electronic behaviors in \(\hbox {CrP}_4\) are mainly dominated by the P-P covalent bonds in \(\hbox {CrP}_4\), so that \(\hbox {CrP}_4\) tend to be a semiconductor due to covalent bonding properties between P-P atoms. While in \(\hbox {MoP}_4\) and \(\hbox {WP}_4\), the electronic properties are largely determined by the metal atoms which have metallic bonds with P atoms, so that they show semimetallic properties. The small peaks on the Fermi level in \(\hbox {MoP}_4\) and \(\hbox {WP}_4\) semimetals are related to the band touching point between the top of valance and the bottom of conduction bands. Similar DOSs around the Fermi level are also found in \(\hbox {CaP}_3\) family of nodal line semimetals41.

Total and partial density of states (DOS) for \(\hbox {MP}_4\) (M = Cr, Mo, W) compounds at equilibrium lattice parameters using HSE06 functional (without spin-orbital coupling). (a, b) Total and partial DOSs for \(\hbox {CrP}_4\); (c, d) Total and partial DOSs for \(\hbox {MoP}_4\); and (e, f) Total and partial DOSs for \(\hbox {WP}_4\).
We have further examined the band structures of \(\hbox {MoP}_{4}\) and \(\hbox {WP}_{4}\) with spin-orbital coupling (SOC) as shown in Fig. S1 in Supplementary Information. For \(\hbox {MoP}_4\), the SOC induced band gap is about 0.1 meV at the \(\Gamma\) point, while for \(\hbox {WP}_4\), the SOC induced band gap is about 29 meV along the high-symmetric X-\(\Gamma\) direction. We can see that when SOC is included, \(\hbox {MoP}_4\) and \(\hbox {WP}_4\) become strong topological insulators with the symmetry-based indicators64,65,66 (\(z_2\), \(z_2\), \(z_2\), \(z_4\)) as (0,0,0,1), like as the finding in \(\hbox {CaP}_3\) family of materials41.
In order to better understand the electronic properties of VIB-\(\hbox {MP}_4\) (\(\hbox {M}= \hbox {Cr}\), Mo, W) compounds, we have also examined the electronic properties of the \(\hbox {VP}_4\), \(\hbox {NbP}_4\), \(\hbox {TaP}_4\), \(\hbox {MnP}_4\), \(\hbox {TcP}_4\) and \(\hbox {ReP}_4\), while V, Nb and Ta are in the VB group, and Mn, Tc and Re are in the VIIB group, which are all next to Cr, Mo and W in the Periodic Table of Elements. The \(\hbox {TcP}_4\) and \(\hbox {ReP}_4\) are experimentally synthesized by the reaction of their constituent elements67,68,69. The calculated equilibrium lattice parameters and electronic band structures are given in Table S1 and Fig. S2 in Supplementary Information, respectively. The structural parameters and electronic behavior that is, \(\hbox {VP}_4\) is metallic and \(\hbox {MnP}_4\) is a semiconductor reported by Gong et al.58. We find that \(\hbox {VB-MP}_4\) (\(\hbox {M}= \hbox {V}\), Nb, Ta) have metallic behavior, while VIIB-\(\hbox {MP}_4\) (\(\hbox {M}= \hbox {Mn}\), Tc, Re) are semiconductors. It is clearly seen that from \(\hbox {VB-MP}_4\) to VIIB-\(\hbox {MP}_4\), the metallicity of these phosphides grow weaker with a change from metallic to semiconducting, while from top (3d) to bottom (5d) in each group, the metallicity of these phosphides grow stronger. So it is reasonable that \(\hbox {CrP}_4\) should be a semiconductor, \(\hbox {MoP}_4\) is a semimetal with isolated nodal points and \(\hbox {WP}_4\) is a topological nodal line semimetal with a line of nodes.
Conclusions
In conclusion, we have performed a systematic ab initio study on \(\hbox {MP}_4\) (\(\hbox {M} = \hbox {Cr}\), Mo, W) monoclinic compounds. Their dynamical stabilities have been confirmed by phonon modes calculations. Electron band structures calculations show that \(\hbox {CrP}_4\) is an indirect band gap semiconductor with a narrow band gap of 0.47 eV, \(\hbox {MoP}_4\) is Dirac semimetal and \(\hbox {WP}_4\) is considered as a new candidate for topological nodal line semimetal with a closed nodal ring in the first BZ protected by the PT symmetry. The electronic density of states calculations indicate that in \(\hbox {CrP}_4\), the valence and conduction bands near the Fermi level are mainly contributed by the p orbitals of P atoms, while in \(\hbox {MoP}_4\) and \(\hbox {WP}_4\), there is a little peak on the Fermi level and the energy bands are mainly composed of d orbitals of Mo and W atoms, respectively. We also make a comparison of the phosphides with group VB and VIIB transition metals and a trend of change from metallic to semiconducting is observed from \(\hbox {VB-MP}_4\) to VIIB-\(\hbox {MP}_4\) compounds. These results provide a systematic understanding and pave the way for further experimental explorations on the transition metal phosphides.
Methods
Our calculations were carried out using the density functional theory as implemented in the Vienna ab initio simulation package (VASP)70. The projector augmented wave (PAW)71 method was adopted with valence electrons of \(3s^23p^3\) for P, \(3p^63d^54s^1\) for Cr, \(4p^64d^55s^1\) for Mo, and \(5p^65d^46s^1\) for W. Generalized gradient approximation (GGA) developed by Perdew, Burke and Ernzerhof (PBE)72 is used as the exchange-correlation potential. A \(5 \times 8 \times 6\) Monkhorst-Pack grid of BZ sampling is used and an energy cutoff of 500 eV is set for the plane-wave basis. The structures are fully optimized until the total energy difference is less then 10\(^{-6}\) eV and convergence criteria for atomic forces is set to be 10\(^{-3}\) eV/Å. The electronic properties are calculated with the Heyd–Scuseria–Ernzerhof hybrid functional (HSE06)60 and the phonon properties are calculated with phononpy package73. To further explore the topological electronic properties, we establish a tight binding (TB) model using the maximally localized Wannier functions (MLWFs)62,63 implemented in Wannier90 package74 and searched the band crossing points in the entire BZ with WannierTools pacakge75.
References
Whitmire, K. H. & Caudell, J. B. First row transition metal phosphides, in encyclopedia of inorganic and bioinorganic chemistry. John Wiley Sons Ltd1–8, https://doi.org/10.1002/9781119951438.eibc2160 (2013).
Chen, W. F., Muckerman, J. T. & Fujita, E. Recent developments in transition metal carbides and nitrides as hydrogen evolution electrocatalysts. Chem. Commun.49, 8896–8909. https://doi.org/10.1039/C3CC44076A (2013).
Veillard, A. Ab initio calculations of transition-metal organometallics: structure and molecular properties. Chem. Rev.91, 743–766. https://doi.org/10.1021/cr00005a006 (1991).
Feng, L. & Huaiguo, X. advances of transition metal phosphide application in electrochemical energy storage and catalysis. ChemElectroChem.4, 20–34. https://doi.org/10.1002/celc.201600563 (2017).
Jing, H. C. & Kenton, H. W. A structural survey of the binary transition metal phosphides and arsenides of the d-block elements. Coord. Chem. Rev.355, 271–327. https://doi.org/10.1016/j.ccr.2017.08.029 (2018).
Du, H., Gu, S., Liu, R. & Li, C. M. Highly active and inexpensive iron phosphide nanorods electrocatalyst towards hydrogen evolution reaction. Int. J. Hydrog Energy40, 14272–14278. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2015).
Carenco, S., Portehault, D., Boissiere, C., Mezailles, N. & Sanchez, C. Nanoscaled metal borides and phosphides: recent developments and perspectives. Chem. Rev.113, 7981–8065. https://doi.org/10.1021/cr400020d (2013).
Yang, S., Liang, C. & Prins, R. Preparation and hydrotreating activity of unsupported nickel phosphide with high surface area. J. Catal.241, 465–469. https://doi.org/10.1016/j.jcat.2006.05.014 (2006).
Muetterties, E. L. & Sauer, J. C. Catalytic properties of metal phosphides: I. Qualitative assay of catalytic properties. J. Am. Chem. Soc.96, 3410–3415. https://doi.org/10.1021/ja00818a012 (1974).
Son, C. Y., Kwak, I. H., Lim, Y. R. & Park, J. FeP and \(\text{FeP}_2\) nanowires for efficient electrocatalytic hydrogen evolution reaction. Chem. Commun.52, 2819–2822. https://doi.org/10.1039/C5CC09832G (2016).
Liu, M. & Li, J. Cobalt phosphide hollow polyhedron as efficient bifunctional electrocatalysts for the evolution reaction of hydrogen and oxygen. J. ACS. Appl. Mater. Interfaces8, 2158–2165. https://doi.org/10.1039/C3CC44076A (2016).
Wu, T., Pi, M., Wang, X., Zhang, D. & Chen, S. Three-dimensional metalrganic framework derived porous \(\text{CoP}_3\) concave polyhedrons as superior bifunctional electrocatalysts for the evolution of hydrogen and oxygen. Phys. Chem. Chem. Phys.19, 2104–2110. https://doi.org/10.1039/C3CC44076A (2017).
Pu, Z., Amiinu, I. S., Wang, M., Yang, Y. & Mu, S. Semimetallic \(\text{MoP}_2\): an active and stable hydrogen evolution electrocatalyst over the whole pH range. Nanoscale8, 8500–8504. https://doi.org/10.1039/C3CC44076A (2016).
Xiao, P. et al. Molybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction. Energy Environ. Sci.8, 2624–2629. https://doi.org/10.1039/C3CC44076A (2015).
Shi, Y. & Zhang, B. Recent advances in transition metal phosphide nanomaterials: synthesis and applications in hydrogen evolution reaction. Chem. Soc. Rev.6, 1529–1541. https://doi.org/10.1039/C5CS00434A (2016).
Dmitruk, N. L., Zuev, V. A. & Stepanova, M. A. Spectral distribution of the photoconductivity of cadmium diphosphide. Russ. Phys. J.34, 642–644. https://doi.org/10.1039/C3CC44076A (1991).
Lazarev, V. B., Shevchenko, V. Y., Grinberg, L. K. & Sobolev. V. V. Semiconducting II-V Compounds. Nauka Moscow, Russia, (1976).
Morozova, V. A., Marenkin, S. F., Koshelev, O. G. & Trukhan, V. M. Optical absorption in monoclinic zinc diphosphide. Inorg. Mater.42, 221–225. https://doi.org/10.1039/C3CC44076A (2006).
Oyama, S. T., Gott, T., Zhao, H. & Lee, Y. K. Transition metal phosphide hydroprocessing catalysts: a review. Catal. Today143, 94–107. https://doi.org/10.1039/C3CC44076A (2009).
Xu, N. et al. Observation of Weyl nodes and Fermi arcs in tantalum phosphide. Nat. Commun.7, 11006–11013. https://doi.org/10.1039/C3CC44076A (2016).
Ceren, T. & Mehmet, C. Electronic structure, phonon and superconductivity for WP 5\(d\)-transition metal. J. Appl. Phys.126, 175103. https://doi.org/10.1039/C3CC44076A (2019).
Autès, G., Gresch, D., Troyer, M., Soluyanov, A. A. & Yazyev, V. O. Robust type-II Weyl semimetal phase in transition metal diphosphides \(\text{XP}_2\) (X = Mo, W). Phys. Rev. Lett.117, 066402. https://doi.org/10.1039/C3CC44076A (2016).
Armitage, N. P., Mele, E. J., Vishwanath, A. & Vishwanath, A. Weyl and Dirac semimetals in three-dimensional solids. Rev. Mod. Phys.90, 015001. https://doi.org/10.1021/cr00005a006 (2018).
Burkov, A. A., Hook, M. D. & Balents, L. Topological nodal semimetals. Phys. Rev. B84, 235126. https://doi.org/10.1021/cr00005a006 (2011).
Volovik, G. E. Quantum phase transitions from topology in momentum space. In quantum analogues: from phase transitions to black holes and cosmology. Phys. Scr.164, 31–73. https://doi.org/10.1021/cr00005a006 (2007).
Wang, J. T., Weng, H. & Chen, C. F. Topological nodal line semimetals in graphene network structures. Adv Phys-X4, 1625724. https://doi.org/10.1021/cr00005a006 (2019).
Jin, H., Su, X. Y., Ni, N. & Zhi, M. Q. Transport of topological semimetals. Annu. Rev. Mater. Res.49, 207–252. https://doi.org/10.1021/cr00005a006 (2019).
Heng, G., Jörn, V. . W. . F. ., Youngkuk, K. . & Andrew, R. . M. . Topological semimetals from first-principles. Annu. Rev. Mater. Res.49, 153–183. https://doi.org/10.1021/cr00005a006 (2019).
Mondal, C., Barman, C. K., Kumar, S., Alam, A. & Pathak, B. Emergence of topological insulator and nodal line semi-metal states in XX’Bi (X= Na, K, Rb, Cs; X’= Ca, Sr). Sci. Rep.9, 1–8. https://doi.org/10.1038/s41598-018-36869-0 (2019).
Huang, H., Liu, J., Vanderbilt, D. & Duan, W. Topological nodal-line semimetals in alkaline-earth stannides, germanides, and silicides. Phys. Rev. B93, 201114. https://doi.org/10.1021/cr00005a006 (2016).
Wang, Z. et al. Dirac semimetal and topological phase transitions in \(\text{A}_3\)Bi (A= Na, K, Rb). Phys. Rev. B85, 195320. https://doi.org/10.1021/cr00005a006 (2012).
Weng, H., Fang, C., Fang, Z., Bernevig, B. A. & Dai, X. Weyl semimetal phase in noncentrosymmetric transition-metal monophosphides. Phys. Rev. X5, 011029. https://doi.org/10.1021/cr00005a006 (2015).
Wan, X., Turner, A. M., Vishwanath, A. & Savrasov, S. Y. Topological semimetal and Fermi-arc surface states in the electronic structure of pyrochlore iridates. Phys. Rev. B83, 205101. https://doi.org/10.1021/cr00005a006 (2011).
Fang, C., Weng, H., Dai, X. & Fang, Z. Topological nodal line semimetals. Chin. Phys. B25, 117106. https://doi.org/10.1002/celc.201600563 (2016).
Kim, Y., Wieder, B. J., Kane, C. L. & Rappe, A. M. Dirac line nodes in inversion-symmetric crystals. Phys. Rev. Lett.115, 036806. https://doi.org/10.1002/celc.201600563 (2015).
Hyart, T., Ojajärvi, R. & Heikkilä, T. T. Two topologically distinct Dirac-line semimetal phases and topological phase transitions in rhombohedrally stacked honeycomb lattices. J. Low Temp. Phys.191, 35–48. https://doi.org/10.1002/celc.201600563 (2018).
Liu, Z. K. et al. Discovery of a three-dimensional topological Dirac semimetal, \(\text{Na}_3\)Bi. Science343, 864–867. https://doi.org/10.1002/celc.201600563 (2014).
Lv, B. et al. Observation of Fermi-arc spin texture in TaAs. Phys. Rev. Lett.115, 217601. https://doi.org/10.1002/celc.201600563 (2015).
Hasan, M. Z. & Kane, C. L. Colloquium: Topological insulators. Rev. Mod. Phys.83, 3045. https://doi.org/10.1002/celc.201600563 (2011).
Qi, X. L. & Zhang, S. C. Topological insulators and superconductors. Rev. Mod. Phys.83, 1057. https://doi.org/10.1002/celc.201600563 (2011).
Xu, Q., Yu, R., Fang, Z., Dai, X. & Weng, H. Topological nodal lines semimetals in the \(\text{CaP}_3\) family of materials. Phys. Rev. B95, 045136. https://doi.org/10.1002/celc.201600563 (2017).
Chan, Y. H., Chiu, C. K., Chou, M. Y. & Schnyder, A. P. \(\text{Ca}_3\text{P}_2\) and other topological semimetals with line nodes and drumhead surface states. Phys. Rev. B93, 205132. https://doi.org/10.1002/celc.201600563 (2016).
Dong, X., Hu, M., He, J. L., Tian, Y. J. & Wang, H. T. A new phase from compression of carbon nanotube with anisotropic Dirac fermions. Sci. Rep.5, 1–7. https://doi.org/10.1002/celc.201600563 (2015).
Feng, X. et al. Monoclinic \(\text{C}_{16}\): \(sp^2\)-\(sp^3\) hybridized nodal-line semimetal protected by \(PT\)-symmetry. Carbon127, 527–532. https://doi.org/10.1016/j.ccr.2017.08.029 (2018).
Cheng, Y., Du, J., Melnik, R., Kawazoe, Y. & Wen, B. Novel three dimensional topological nodal line semimetallic carbon. Carbon98, 468–473. https://doi.org/10.1016/j.ccr.2017.08.029 (2016).
Weng, H. et al. Topological node-line semimetal in three-dimensional graphene networks. Phys. Rev. B92, 045108. https://doi.org/10.1016/j.ccr.2017.08.029 (2015).
Wang, J. T. et al. Body-centered orthorhombic \(\text{C}_{16}\): A novel topological node-line semimetal. Phys. Rev. Lett.116, 195501. https://doi.org/10.1016/j.ccr.2017.08.029 (2016).
Cheng, Y. et al. Body-centered tetragonal \(\text{C}_{16}\): A novel topological node-line semimetallic carbon composed of Tetrarings. Small13, 1602894–1602901. https://doi.org/10.1016/j.ccr.2017.08.029 (2017).
Chen, Y. et al. Nanostructured carbon allotropes with Weyl-like loops, points. Nano Lett.15, 6974–6978. https://doi.org/10.1016/j.ccr.2017.08.029 (2015).
Li, Z. Z. et al. Orthorhombic carbon \(\text{oC}_{24}\): A novel topological nodal line semimetal. Carbon133, 39–43. https://doi.org/10.1016/j.ccr.2017.08.029 (2018).
Wang, J. T., Chen, C. F. & Kawazoe, Y. Topological nodal line semimetal in an orthorhombic graphene network structure. Phys. Rev. B97, 245147. https://doi.org/10.1016/j.ccr.2017.08.029 (2018).
Wang, J. T., Qian, Y., Weng, H., Wang, E. G. & Chen, C. F. Three-dimensional crystalline modification of graphene in all-\(sp^2\) hexagonal lattices with or without topological nodal lines. J. Phys. Chem. Lett.10, 2515–2521. https://doi.org/10.1016/j.ccr.2017.08.029 (2019).
Wang, J. T., Nie, S., Weng, H. & Chen, C. F. Topological nodal-net semimetal in a graphene network structure. Phys. Rev. Lett.120, 026402. https://doi.org/10.1016/j.ccr.2017.08.029 (2018).
Bu, K., Wang, J. T., Weng, H. & Chen, C. F. Topological semimetal in an \(sp^2\)-\(sp^3\) hybridized carbon network with nodal rings. Phys. Rev. B101, 205104. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2020).
Jeitschko, W. & Donohue, P. C. The high pressure synthesis, crystal structure, and properties of \(\text{CrP}_4\) and \(\text{MoP}_4\). Acta. Cryst.28, 1893–1898. https://doi.org/10.1016/j.ijhydene.2015.02.099 (1972).
Brown, A. Refinement of the crystal structure of black phosphorus. Acta. Crystallogr.19, 684–685. https://doi.org/10.1016/j.ijhydene.2015.02.099 (1965).
Cheng, Y. et al. Sodium-induced reordering of atomic stacks in black phosphorus. Chem. Mater.29, 1350–1356. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2017).
Gong, N. et al. Structural diversity and electronic properties of 3d transition metal tetraphosphides, \(\text{TMP}_4\) (TM= V, Cr, Mn, and Fe). Inorg. Chem.57, 9385–9392. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2018).
Winkler, B. et al. Crystal chemistry of molybdenum phosphides from density functional theory calculations. J. Phys. Chem. Solids64, 405–411. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2003).
Krukau, A. V., Vydrov, O. A., Izmaylov, A. F. & Scuseria, G. E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys.125, 224106–224112. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2006).
In our calculations, under standard GGA (without spin-orbital coupling) and TB model, \(\text{MoP}_4\) also has a small nodal ring like as \(\text{WP}_4\).
Marzari, N., Mostofi, A. A., Yates, J. R., Souza, I. & Vanderbilt, D. Maximally localized Wannier functions: theory and applications. Rev. Mod. Phys.84, 1419. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2012).
Mostofi, A. A. et al. A tool for obtaining maximally-localised Wannier functions. Comput. Phys. Commun.178, 685–699. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2008).
Po, H. C., Vishwanath, A. & Watanabe, H. Symmetry-based indicators of band topology in the 230 space groups. Nat. Commun.8, 1–9. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2017).
Song, Z., Zhang, T., Fang, Z. & Fang, C. Quantitative mappings between symmetry and topology in solids. Nat. Commun.9, 1–7. https://doi.org/10.1021/cr400020d (2019).
Song, Z., Zhang, T. & Fang, C. Diagnosis for nonmagnetic topological semimetals in the absence of spin-orbital coupling. Phys. Rev. X8, 031069. https://doi.org/10.1021/cr400020d (2018).
Jeitschko, W. & Rühl, R. Synthesis and crystal structure of diamagnetic \(\text{ReP}_4\), a polyphosphide with Re-Re pairs. Acta Crystallogr. B35, 1953–1958. https://doi.org/10.1021/cr400020d (1979).
Rühl, R., Jeitschko, W. & Schwochau, K. Preparation and crystal structures of technetium phosphides. J. Solid State Chem.44, 134–140. https://doi.org/10.1021/cr400020d (1982).
Feng, S., Cheng, X., Cheng, X., Yue, J. & Li, J. Theoretical study on electronic, optical properties and hardness of technetium phosphides under high pressure. Crystals.7, 176–185. https://doi.org/10.1021/cr400020d (2017).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B54, 11169. https://doi.org/10.1021/cr400020d (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B50, 17953. https://doi.org/10.1021/cr400020d (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett.78, 17954. https://doi.org/10.1021/cr400020d (1997).
Togo, A., Oba, F. & Tanaka, I. First-principles calculations of the ferroelastic transition between rutile-type and \(\text{CaCl}_2\)-type \(\text{SiO}_2\) at high pressures. Phys. Rev. B78, 134106. https://doi.org/10.1021/cr400020d (2008).
Mostofi, A. A. et al. An updated version of wannier90: A tool for obtaining maximally-localised Wannier functions. Comp. Phys. Comm.185, 2309–2310. https://doi.org/10.1016/j.ijhydene.2015.02.099 (2014).
Wu, Q., Zhang, S., Song, H. F., Troyer, M. & Soluyanov, A. A. WannierTools: An open-source software package for novel topological materials. Phys. Comm.224, 405–416. https://doi.org/10.1016/j.jcat.2006.05.014 (2018).
Momma, K. & Izumi, F. VESTA: a three-dimensional visualization system for electronic and structural analysis. J. Appl. Cryst.41, 653–658. https://doi.org/10.1016/j.jcat.2006.05.014 (2008).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grants No. 11974387 and No. 11674364) and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB33000000).
Author information
Authors and Affiliations
Contributions
M.R.K. and J.T.W. designed the study and wrote the paper; M.R.K and J.S.C. drawn Fig. 1, M.R.K. calculated the phonon band structures and plot Fig. 2, M.R.K. and K.B. calculated the electronic band structures and plot the Fig. 3, M.R.K. and J.S.C. plot the Fig. 4; all authors discussed the results and contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khan, M.R., Bu, K., Chai, JS. et al. Novel electronic properties of monoclinic MP4 (M = Cr, Mo, W) compounds with or without topological nodal line. Sci Rep 10, 11502 (2020). https://doi.org/10.1038/s41598-020-68349-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-68349-9
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
