
Abstract
Trapping lithium polysulfides (LiPSs) on a material effectively suppresses the shuttle effect and enhances the cycling stability of Li–S batteries. For the first time, we advocate a recently synthesized two-dimensional material, biphenylene, as an anchoring material for the lithium-sulfur battery. The density functional theory calculations show that LiPSs bind with pristine biphenylene insubstantially with binding energy ranging from −0.21 eV to −1.22 eV. However, defect engineering through a single C atom vacancy significantly improves the binding strength (binding energy in the range −1.07 to −4.11 eV). The Bader analysis reveals that LiPSs and S8 clusters donate the charge (ranging from −0.05 e to −1.12 e) to the biphenylene sheet. The binding energy of LiPSs with electrolytes is smaller than those with the defective biphenylene sheet, which provides its potential as an anchoring material. Compared with other reported two-dimensional materials such as graphene, MXenes, and phosphorene, the biphenylene sheet exhibits higher binding energies with the polysulfides. Our study deepens the fundamental understanding and shows that the biphenylene sheet is an excellent anchoring material for lithium-sulfur batteries for suppressing the shuttle effect because of its superior conductivity, porosity, and strong anchoring ability.
Similar content being viewed by others
Introduction
Over the last three decades, lithium-ion rechargeable batteries have gained vast popularity due to their low self-discharge, ample energy storage, stable cycling performance, higher theoretical capacity, and specific energy density, which directly affected the development of energy storage technologies1,2,3. Li-ion batteries are environmental-friendly and prominent for portable electronics, as they offer much higher energy density than other rechargeable systems4,5,6. Furthermore, lithium-sulfur batteries are excellent for next-generation rechargeable batteries with a theoretical capacity of 1675 mAh g−1. and specific energy of 2600 Wh Kg−1, respectively, higher than commercial Li-ion batteries. In addition, the sulfur cathode has many merits of abundant resources, cheap and non-pollution2,7,8. Further advances in rechargeable batteries are essential to fulfill electric vehicles and energy storage demand.
Current research efforts are focused on overcoming the challenges that limit the efficiency of lithium-sulfur (Li–S) battery, such as capacity fade, the solubility of active species, discharge/charge characteristics, and the most notorious and intractable shuttle effect9,10,11. The shuttle effect is the migration of sulfur back and forth between the positive and negative electrodes in the charging and discharging process12,13. The S atoms also react with the lithium anode and form lithium polysulfides (LiPSs) Li2Sx, 2 < x ≤ 8, which leads to corrosion, resulting in fast capacity fading and low Coulombic efficiency14. There are two main methods to suppress the shuttle effect, one is by preventing the diffusion of polysulfides into electrolytes, and the other is by using an anchoring material to block the migration of the polysulfides to the anode15. In the first case, polysulfides are diverted towards a material present in the cathode or electrolyte to decrease the solubility16. The second strategy is carried out by introducing a material to block the diffusion pathway of polysulfides. The diffusion process will be restrained when the binding of LiPSs clusters to the host material is more robust than that to the electrolytes.
A suitable anchoring material should have excellent conductivity, high surface area, porous structure, and high binding energy with the polysulfides17,18, preventing polysulfides from dissolving into electrolytes. Several two-dimensional (2D) materials have been proposed as anchoring materials employed to suppress the shuttling effect, such as graphene, OH, F, and S terminated Ti2C MXene17, porous vanadium nitride nanoribbon with graphene19,20, and boro-phosphorene21. The biphenylene (BPN), a planar fully sp2-hybridized allotrope of carbon, is recently synthesized22 where carbon atoms in BPN are arranged in square, hexagonal, and octagonal rings. It is metallic with a Dirac cone above the Fermi level, suggesting high carrier mobility and good conductivity23. Also, it shows a planar lattice which is one key factor for the design of batteries with a fast charge/discharge rate, so it is expected to exhibit improved performance as a potential anchoring material24. Therefore, the first-principles calculations are carried out to evaluate the potential of 2D BPN sheet as an anchoring material for Li–S batteries.
Computational methods
The first-principles spin-polarized calculations are performed using the Vienna ab-initio Simulation Package (VASP)25. The projector augmented wave method is adopted to describe the ion–electron interactions25, which is treated by the generalized gradient approximation with the Perdew-Burke-Ernzerhof formalism26. The van der Waals interactions are implemented considering the scheme of Grimme (DFT-D3) with zero damping correction27. The plane-wave cutoff kinetic energy is set to 550 eV. The total ground state energy and force convergence criteria is set to 10–6 eV and 0.05 eV/Å, respectively. The interactions between two adjacent periodic images are eliminated by introducing a vacuum of 24 Å perpendicular to the sheet. A 3 × 3 × 1 supercell of BPN with the Monkhorst–Pack k-point mesh of 3 × 3 × 1 is used for geometry optimization and self-consistent calculations. The binding energy (Eb) of LiPSs and S8 clusters with the BPN sheet is calculated using Eb = Etotal − (Ebiphenylene + ELiPSs/S8), where Etotal, Ebiphenylene, and ELiPSs/S8 refer to the total energy of biphenylene adsorbed with Li2Sx/S8, pristine biphenylene, and the isolated Li2Sx/S8 clusters, respectively.
Results and discussion
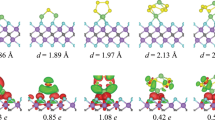
The BPN sheet possesses a carbon lattice with a rectangular primitive unit cell formed by six C atoms having tetragon, hexagon, and octagon rings. The optimized lattice constants of the BPN sheet are 4.51 Å and 3.76 Å, consistent with the previous study24. The electron localization function along the (0 0 1) plane shows the underlying characteristics of C–C bonds (see Fig. 1b) and confirms that a large proportion of bonds in the BPN sheet are strong covalent bonds. In Fig. 1b, the red color shows intense localization and, in turn, a stronger bond. As illustrated in Fig. 1a, there are eight different adsorption sites. The S1, S2, and S3 sites are the octagon, tetragon, and hexagon hollows, respectively, and the S4, S5, and S6 sites are the distinctive bridges between the carbon atoms, and S7, S8 which are randomly chosen carbon atoms. Since lattice defects are inevitable during the synthesis process, the bindings of LiPSs and S8 are also investigated on the defective BPN sheet having a single C vacancy. Such defects can also be generated using a laser/electron beam of an appropriate energy bombardment28. There are two distinctive carbon atoms in the BPN sheet (marked with two dotted circles in Fig. 1a and named them D1 and D2, respectively), leading to two distinctive single vacancy defects. The structures with the single defect are illustrated in Fig. 1c–f combined with their respective electron localization function plots. In both cases, intense red color is noticed at the vacancy site, indicating a free electron (dangling bond), which directly affects the bonding of polysulfides with the BPN sheet. In addition, the defective BPN sheets exhibit a magnetic moment of a value of 1 μB in both D1 and D2 cases, which is directly reflected in the calculated density of states. Compared to pristine BPN, the defective BPN sheets DOS is asymmetrical, which can be explained by the fact that when a vacancy has created the relaxation of its nearest neighbor atoms and resulting charge redistribution saturate the orbitals of two of the three C atoms, and the remaining unsaturated carbon atom is responsible for the magnetic moment. The computed density of states of pristine BPN and defective sheets are available in the supplementary information (SI) (see Fig. S2). Figure S2 shows that the biphenylene sheet is metallic, resulting in better anchoring material than phosphorene. Much higher performance as an anchoring material is expected of its porous nature compared to graphene and MXene.

(a) Crystal structure of pristine BPN sheet with the possible adsorption sites (S1-S8) (b) The electron localization function of pristine BPN sheet (c,e) crystal structure of defective BPN sheet, and (d,f) the corresponding electron localization function.
The strength of the chemical bond in the defective BPN sheet is investigated with the aid of the projected crystal-orbital Hamilton population (-pCOHP) method, in which -pCOHP is divided into bonding and antibonding states, as shown in Fig. 2. In both cases, minor antibonding states near the Fermi level are observed, related to the electronic structure distortion resulting from the lone electron pair, making the two spin sublattices inequivalent. Apart from the minor antibonding, the C–C bonds contribute to the chemical stability of the bonding region. All the filled bands below the Fermi level contribute to chemical stability.

COHP for defected BPN sheet (a) corresponds to D1 defected sheet (b) corresponds to D2 defected sheet. The Fermi level is at zero.
As cathode discharge, polysulfides significantly influence the capacity and cycling stability of Li–S batteries. The optimized most stable molecular structure of Li2Sx (x = 1, 2, 4, 6, 8) and S8 clusters are shown in the supplementary information (Fig. S3 in SI). The Li–S bond is more extended than the S–S bond, indicating that as the number of sulfur atoms increases, the high-order LiPSs can easily be ionized as Li+ and polysulfide anions, resulting in the so-called shuttle effect9. The binding energy of the LiPSs and S8 clusters with the BPN sheet is calculated by considering the most stable adsorption configuration (Fig. S1 in SI). Among the eight different adsorption sites, in most cases, LiPSs preferred to adsorb near the C–C bond of the octagon ring (S6 site in Fig. 1) because of charge accumulation. The maximum binding energy is −1.22 eV for Li2S, whereas the minimum binding energy of −0.43 eV is obtained for Li2S4.
The binding energy decreases gradually with the increase of the S atoms, but it also differs with the orientations of the molecule. When the S atom increased to 2 in Li2S2, the binding energy decreased by 0.342 eV because of the conformational changes noticed in the relaxed configurations. Two lithium atoms of Li2S are facing toward the sheet compared to only one lithium atom in Li2S2, directly resulting in lower binding energy. The highest binding energy values for each cluster with the corresponding favorable adsorption site are listed in Table 1, and the binding energy for each site is presented in the SI (see Fig S4), which ensures the stability of LiPSs on the BPN sheet. However, the binding energy values for the x > 3 are low and need further improvement. The binding energy of polysulfides is enhanced with defected BPN sheet. All the polysulfide clusters are placed at the top of the defect, and then the structures are relaxed, as shown in Figs. 3 and 4. The single C vacancy shows high charge localization (Fig. 1), shared with the Li2Sx and S8 clusters, resulting in higher binding energy than the pristine BPN sheet. For all Li2Sx clusters except with Li2S6 in defected BPN sheet, two Li atoms lie nearest to the surface. For Li2S6, only one Li atom faces the surface, leading to the decreased binding energy. The S8 cluster is positioned at 1.73 Å from both defective surfaces, compared with a 3.49 Å in the pristine surface results increased binding energy of S8 from −0.21 eV (pristine BPN sheet) to −1.59 eV in the defective case. The minimum distances between the LiPSs clusters and the defective BPN sheets range from 1.67 Å (Li2S8) to 2.18 Å (Li2S6).

Ground state configuration of defective BPN sheet, (D1) adsorbed with (a) Li2S (b) Li2S2 (c) Li2S4 (d) Li2S6 (e) Li2S8 and (f) S8 clusters. The brown, green, and yellow spheres correspond to C, Li, and S atoms, respectively.

Ground state configuration of defective BPN sheet (D2) adsorbed with (a) Li2S (b) Li2S2 (c) Li2S4 (d) Li2S6 (e) Li2S8 and (f) S8 clusters. The brown, green, and yellow spheres correspond to C, Li, and S atoms, respectively.
The leading cause of the shuttle effect in Li–S batteries is the smaller binding energy of LiPSs and S8 clusters with the cathode surface than with the electrolyte. Therefore, dissolution in the electrolytes and diffusion to the anode of LiPSs is easy. The binding energies of LiPSs with (1,3-dioxolane (DOL) and 1,2-dimethoxyethane (DME)) electrolytes are calculated. The binding energies of LiPSs with graphene are also listed to gain a more profound sense of BPN sheet as an anchoring material. The binding energies of LiPSs with DME and DOL molecules are smaller than those with the defected BPN sheet and higher than those with graphene, which indicates that graphene could not effectively trap LiPSs, but the defected BPN sheet can effectively trap LiPSs. Compared with other studied anchoring materials such as MXene (−0.83 eV to −4.31 eV)17, phosphorene (−0.75 eV to −2.51 eV)29, porous carbon allotropes (T-graphene (T-G) (−1.37 eV to −2.71 eV), Popgraphene (P-G) (−1.18 eV to e −1.91 eV ), Dodecagraphene (D-G) (−0.525 to −1.334), R-graphyne (R-G) (−0.735 to −1.009)30), the binding energy of LiPSs with the defected BPN sheet is more substantial, which indicates that the BPN sheet is indeed a promising anchoring material.
Defected BPN sheet outperforms the pristine sheet in the binding energy of LiPSs and S8 clusters. The Bader analysis shows that the LiPSs and S8 donate the charge of between −0.05 e to −1.02 e to the BPN sheet (see Table 2). The charge is mainly accumulated (depleted) on S (Li) before absorption and transferred substantially to the sheet after absorption of clusters. The charge density difference between LiPSs and the defective BPN sheet is calculated according to \(\Delta \rho ={\rho }_{total}-{\rho }_{defectiveBPN}-{\rho }_{LiPSs/S_{8}}\), where \({\rho }_{total}\), \({\rho }_{defectiveBPN}\), and \({\rho }_{LiPSs/S_{8}}\) are the calculated charge density of combined BPN sheet and LiPSs/S8 systems, defected BPN sheet alone, and the isolated LiPSs/S8 clusters, respectively. The calculated charge density difference of the defective BPN sheet is shown in Figs. 5 and 6.

Calculated charge density difference of defective BPN sheet (D1 site) adsorbed with (a) Li2S, (b) Li2S2, (c) Li2S4, (d) Li2S6, (e) Li2S8, and (f) S8. Yellow and blue regions indicate charge accumulation and charge depletion, respectively, the iso-surface is set to 0.002 e/A3. The brown, green, and yellow spheres correspond to C, Li, and S atoms, respectively.

Calculated charge density difference of defective BPN sheet (D2 site) adsorbed with (a) Li2S, (b) Li2S2, (c) Li2S4, (d) Li2S6, (e) Li2S8, and (f) S8. Yellow and blue regions indicate charge accumulation and depletion, respectively, the iso-surface value is 0.002 e/A3. The brown, green, and yellow spheres correspond to C, Li, and S atoms, respectively.
Figure 5 shows, Li2S6 has the least apparent charge redistribution compared to the other clusters. For Li2Sx (x = 1, 2, 4, 8) and S8 adsorption, electrons are mainly accumulated in the region between Li and S nearest. The detailed charge rearrangement on the constituents of LiPSs/S8 Species before and after adsorption on BPN sheet are listed in the SI (Table S1). Li2S positioned itself at 1.71 Å from the defective BPN sheet in the D1 case, where two lithium atoms facing the surface led to the highest charge transferred value. Moreover, most carbon materials have low dipole moments, while Li2S and Li2S2 have high dipole moments. When the BNP sheet interacts with Li2S and Li2S2, a strong dipole–dipole electrostatic interaction results in the highest charge transfer between them34,35. On the contemporary, Li2S6 has the lowest charge transferred within the lithium polysulfides and its highest distance, 2.18 Å, on top of the D1 defective BPN sheet.
Conclusion
In summary, the density functional theory calculations are carried out to explore the potential and performance of 2D BPN sheet as an anchoring material for Li–S batteries. The results show that the defective BPN sheet significantly improves the binding of LiPSs with binding energies ranging from −1.07 to −4.11 eV, which can effectively inhibit the shuttle effect and reduce the migration barrier to realize the rapid realization charge/discharge. The Bader charge analysis shows an electronic charge transfer ranging from −0.05 e to −1.12 e from LiPSs to the BPN sheet. Compared to graphene, MXenes, and other potential anchoring materials, the BPN sheet has the highest binding energies, making it a more efficient and reliable choice. Our work deepens the fundamental understanding of the anchoring mechanism and demonstrate that the biphenylene sheet has excellent potential to be an outstanding anchoring material for Li–S batteries for suppressing the shuttle effect.
Data availability
No datasets were generated or analysed during the current study.
References
Zhou, L., Danilov, D. L., Eichel, R. A. & Notten, P. H. L. Host materials anchoring polysulfides in Li–S batteries reviewed. Adv. Energy Mater. 11(15), 1. https://doi.org/10.1002/aenm.202001304 (2021).
Bruce, P. G., Freunberger, S. A., Hardwick, L. J. & Tarascon, J.-M. Li–O2 and Li–S batteries with high energy storage. Nat. Mater. 11(02), 1 (2011).
Chung, W. J. et al. The use of elemental sulfur as an alternative feedstock for polymeric materials. Nat. Chem. 5(6), 1. https://doi.org/10.1038/nchem.1624 (2013).
Manthiram, A.; Fu, Y.; Chung, S.; Zu, C.; Su, Y. Rechargeable Lithium − Sulfur Batteries.
Crabtree, G. Perspective: The energy-storage revolution. Nature https://doi.org/10.1038/526S92a (2015).
Budde-Meiwes, H., Drillkens, J., Lunz, B., Muennix, J., Rothgang, S., Kowal, J., Sauer, D. U. A review of current automotive battery technology and future prospects. Proc. Inst. Mech. Eng. Part D J. Automobile Eng. (2013)
Yang, Y., Zheng, G. & Cui, Y. Nanostructured sulfur cathodes. Chem. Soc. Rev. 42(7), 1. https://doi.org/10.1039/c2cs35256g (2013).
Gao, X. P. & Yang, H. X. Multi-electron reaction materials for high energy density batteries. Energy Environ. Sci. https://doi.org/10.1039/b916098a (2010).
Wild, M. et al. Lithium sulfur batteries, a mechanistic review. Energy Environ. Sci. 8(12), 3477–3494. https://doi.org/10.1039/c5ee01388g (2015).
Diao, Y., Xie, K., Xiong, S. & Hong, X. Shuttle phenomenon-the irreversible oxidation mechanism of sulfur active material in Li-S battery. J. Power Sources 235, 1. https://doi.org/10.1016/j.jpowsour.2013.01.132 (2013).
Fang, R. et al. More reliable lithium-sulfur batteries: Status, solutions and prospects. Adv. Mater. https://doi.org/10.1002/adma.201606823 (2017).
Xiong, S., Xie, K., Diao, Y. & Hong, X. Characterization of the solid electrolyte interphase on lithium anode for preventing the shuttle mechanism in lithium-sulfur batteries. J. Power Sources 246, 1. https://doi.org/10.1016/j.jpowsour.2013.08.041 (2014).
Xu, G. et al. High performance lithium-sulfur batteries: Advances and challenges. J. Mater. Chem. A. https://doi.org/10.1039/c4ta02097a (2014).
Peng, H. J. & Zhang, Q. Designing host materials for sulfur cathodes: From physical confinement to surface chemistry. Angew. Chemie - Int. Ed. 54(38), 11018–11020. https://doi.org/10.1002/anie.201505444 (2015).
Ren, W., Ma, W., Zhang, S. & Tang, B. Recent advances in shuttle effect inhibition for lithium sulfur batteries. Energy Storage Mater. 23, 707–732. https://doi.org/10.1016/j.ensm.2019.02.022 (2019).
Wu, D. S. et al. Quantitative investigation of polysulfide adsorption capability of candidate materials for Li-S batteries. Energy Storage Mater. 13, 1. https://doi.org/10.1016/j.ensm.2018.01.020 (2018).
Liu, X., Shao, X., Li, F. & Zhao, M. Anchoring effects of S-terminated Ti 2 C MXene for Lithium–Sulfur batteries: A first-principles study. Appl. Surf. Sci. 455, 522–526. https://doi.org/10.1016/j.apsusc.2018.05.200 (2018).
Li, Z., Zhang, J. & Lou, X. W. Hollow carbon nanofibers filled with MnO2 nanosheets as efficient sulfur hosts for lithium-sulfur batteries. Angew. Chemie - Int. Ed. 54(44), 1. https://doi.org/10.1002/anie.201506972 (2015).
Sun, Z. et al. Conductive porous vanadium nitride/graphene composite as chemical anchor of polysulfides for lithium-sulfur batteries. Nat. Commun. 8, 1. https://doi.org/10.1038/ncomms14627 (2017).
Li, Y. et al. A MnO2/graphene oxide/multi-walled carbon nanotubes-sulfur composite with dual-efficient polysulfide adsorption for improving lithium-sulfur batteries. ACS Appl. Mater. Interfaces 8(42), 1. https://doi.org/10.1021/acsami.6b04270 (2016).
Zhang, H. et al. Borophosphene: A potential anchoring material for lithium-sulfur batteries. Appl. Surf. Sci. 562(May), 150157. https://doi.org/10.1016/j.apsusc.2021.150157 (2021).
Matthias, R., Stefan, R. & Michael, J. Biphenylene Network : A Nonbenzenoid Carbon Allotrope. https://doi.org/10.1126/science.abg4509 (2021).
Bafekry, A. et al. Biphenylene monolayer as a two-dimensional nonbenzenoid carbon allotrope: A first-principles study. J. Phys. Condens. Matter 1, 1–5. https://doi.org/10.1088/1361-648x/ac2a7b (2021).
Bafekry, A. et al. Biphenylene monolayer as a two-dimensional nonbenzenoid carbon allotrope: A first-principles study. J. Phys. Condens. Matter https://doi.org/10.1088/1361-648x/ac2a7b (2021).
Dupuy, M. S. Projector Augmented-Wave Method: An Analysis in a One-Dimensional Setting. ESAIM Math. Model. Numer. Anal. 54(1), 1. https://doi.org/10.1051/m2an/2019017 (2020).
Perdew, J. P., Burke, K., Ernzerhof, M. P. & Burke, E. Generalized gradient approximation made simple(2). Phys. Rev. Lett. 77(3), 1 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate Ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132(15), 154104. https://doi.org/10.1063/1.3382344 (2010).
Tian, W., Li, W., Yu, W. & Liu, X. A review on lattice defects in graphene: Types generation effects and regulation. Micromachines. https://doi.org/10.3390/mi8050163 (2017).
Zhao, J., Yang, Y., Katiyar, R. S. & Chen, Z. Phosphorene as a promising anchoring material for lithium-sulfur batteries: A computational study. J. Mater. Chem. A 4(16), 6124–6130. https://doi.org/10.1039/c6ta00871b (2016).
Li, T., He, C. & Zhang, W. Rational design of porous carbon allotropes as anchoring materials for lithium sulfur batteries. J. Energy Chem. 52, 121–129. https://doi.org/10.1016/j.jechem.2020.04.042 (2021).
Jiang, H. R., Shyy, W., Liu, M., Ren, Y. X. & Zhao, T. S. Borophene and defective borophene as potential anchoring materials for lithium-sulfur batteries: A first-principles study. J. Mater. Chem. A 6(5), 1. https://doi.org/10.1039/c7ta09244j (2018).
Wang, B., Alhassan, S. M. & Pantelides, S. T. Formation of large polysulfide complexes during the lithium-sulfur battery discharge. Phys. Rev. Appl. 2(3), 1. https://doi.org/10.1103/PhysRevApplied.2.034004 (2014).
Yin, L. C. et al. Understanding the interactions between lithium polysulfides and N-doped graphene using density functional theory calculations. Nano Energy 25, 1. https://doi.org/10.1016/j.nanoen.2016.04.053 (2016).
Zhang, Q. et al. Understanding the anchoring effect of two-dimensional layered materials for lithium-sulfur batteries. Nano Lett. 15(6), 1. https://doi.org/10.1021/acs.nanolett.5b00367 (2015).
Hou, T. Z. et al. Design principles for heteroatom-doped nanocarbon to achieve strong anchoring of polysulfides for lithium-sulfur batteries. Small https://doi.org/10.1002/smll.201600809 (2016).
Acknowledgements
N. S. acknowledge the financial support from Khalifa University of Science and Technology under the startup grant FSU-2020-11/2020. Authors acknowledge the contribution of Khalifa University's high-performance computing and research computing facilities to the results of this research.
Author information
Authors and Affiliations
Contributions
H.K.Al-J. performed the calculations and wrote the original draft. M.S. contributes to the calculations and in writing the manuscript. N.S. and K.L create the idea and supervise the project and finally review the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Al-Jayyousi, H.K., Sajjad, M., Liao, K. et al. Two-dimensional biphenylene: a promising anchoring material for lithium-sulfur batteries. Sci Rep 12, 4653 (2022). https://doi.org/10.1038/s41598-022-08478-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-08478-5
This article is cited by
-
PHOTH-graphene: a new 2D carbon allotrope with low barriers for Li-ion mobility
Scientific Reports (2024)
-
Colossal figure of merit and compelling HER catalytic activity of holey graphyne
Scientific Reports (2023)
-
Investigating the pinhole effect on the mechanical properties of biphenylene
Applied Physics A (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
