
Abstract
We identified two NEXMIF variants in two unrelated individuals with non-autoimmune diabetes and autistic traits, and investigated the expression of Nexmif in mouse and human pancreas and its function in pancreatic beta cells in vitro and in vivo. In insulin-secreting INS-1E cells, Nexmif expression increased strongly in response to oxidative stress. CRISPR Cas9-generated Nexmif knockout mice exhibited a reduced number of proliferating beta cells in pancreatic islets. RNA sequencing of pancreatic islets showed that the downregulated genes in Nexmif mutant islets are involved in stress response and the deposition of epigenetic marks. They include H3f3b, encoding histone H3.3, which is associated with the regulation of beta-cell proliferation and maintains genomic integrity by silencing transposable elements, particularly LINE1 elements. LINE1 activity has been associated with autism and neurodevelopmental disorders in which patients share characteristics with NEXMIF patients, and can cause genomic instability and genetic variation through retrotransposition. Nexmif knockout mice exhibited various other phenotypes. Mortality and phenotypic abnormalities increased in each generation in both Nexmif mutant and non-mutant littermates. In Nexmif mutant mice, LINE1 element expression was upregulated in the pancreas, brain, and testis, possibly inducing genomic instability in Nexmif mutant mice and causing phenotypic variability in their progeny.
Similar content being viewed by others

Introduction
The human neurite extension and migration factor (NEXMIF) is located on the X-chromosome at locus Xq13.3 in a genomic region of 192 kb that is associated with intellectual disabilities as well as other phenotypes in humans1. The X chromosome is highly enriched with genes that are expressed in the brain, and these include NEXMIF2,3. NEXMIF orthologs are highly conserved in mammals, and even in chicken and zebrafish a substantial similarity is observed (61% and 32% sequence identity, respectively). However, at the protein level most of the similarity between human and zebrafish Nexmif is absent, with the exception of 112 amino acids in a region of about 250 amino acids (amino acids 310–598)4. This 112-amino acid sequence is conserved in all species expressing Nexmif and is similar to a small portion of the amino acid sequence of the polymerase zeta subunit REV3L2,5. DNA polymerase zeta is a low-fidelity DNA polymerase that has an important role in maintaining genomic stability and controlling mutagenesis6,7. Further, it has been shown that Nexmif is highly expressed in the neurons of the developing mouse brain, where it contributes to the regulation of neurite outgrowth3,8. Neurons and beta cells share similar signal transduction pathways and several transcription factors. Several of these transcription factors have been found to be mutated in patients with both mental retardation or brain anomalies and diabetes9,10,11,12,13. Based on its expression pattern in the mouse brain and association with mental retardation, NEXMIF has been suggested to have a function similar to that of ATRX, which is involved in chromatin modeling and the maintenance of genomic stability by the silencing of repetitive elements14. This, together with the amino acid sequence similarity to REV3L, suggests that NEXMIF might play a role in maintaining genomic stability. Here we performed in vitro experiments using INS-1E beta cells and in vivo investigations on a Nexmif knockout mouse model to determine the role of Nexmif.
Results
Identification of two subjects with a neurodevelopmental syndrome, a NEXMIF variant, and diabetes
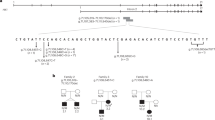
Case 1
The patient, a 15-year-old boy, was born to nonconsanguineous parents of Portuguese origin, at 37 weeks of gestation with a birth weight of 3190 g (P50-P75) and a length of 50 cm (P50). The mother developed gestational diabetes during pregnancy and was treated with insulin. At the age of 4 months, hypotonia was observed, and at 6 months retarded motor development was noticed. At the age of 13 months a first epileptic seizure was seen and treatment with valproic acid was started and pursued until the age of 2 years. Cerebral MRI was within normal limits. A genetic analysis done for severe intellectual disability (IQ 20–34) indicated a 46, XY karyotype, and an array comparative genomic hybridization (aCGH) revealed no duplication or deletion. We identified the pathogenic hemizygous NEXMIF variant c.652C>T, p.Arg218* (NM_001008537.3). At the age of 5 years, diabetes was diagnosed after 3 weeks of polyuria and polydipsia with a postprandial glycemia of 26 mmol/L, a capillary beta-hydroxybutyrate level of 0.7 mmol/L (normal < 0.6 mmol/L) and an HbA1c of 91 mmol/mol (10.5%). Autoimmune autoantibodies such as GADA, IAA, IA2A, ZnT8A, and ICA were all negative. The insulin dose was increased gradually from 0.2 to 0.9 U/kg/day. At the age of 15 years the patient is still nonverbal, with autistic behavior.
Case 2
The second subject, a 30-year-old women, harbors the pathogenic heterozygous NEXMIF c.3470C>A, p.Ser1157* variant associated with mild learning disability, language impairment, epilepsy, and autistic features15. She developed autoimmune antibody-negative (GAD and IA2) diabetes at the age of 28 years. Her glucose level at diagnosis was 18.4 mmol/L, with a concomitant capillary beta-hydroxybutyrate level of 3.7 mmol/L. She was first treated with insulin, which could gradually be replaced by an oral antidiabetic treatment with metformin (2 × 1000 mg/day) and gliclazide modified release (2 × 60 mg/day). Her BMI was 34 kg/m2 at that time. Her 9-year-old son carries the same NEXMIF variant, with severe mental retardation, absence of language, and autistic behavior. The boy has no dysglycemia to date.
Nexmif is expressed in beta cells
As Nexmif was previously described to be highly expressed in the mouse and human brain2 (see GTExportal (https://gtexportal.org/home/gene/KIAA2022, accessed Dec 15, 2019), we used brain tissue as a positive control. Nexmif was indeed expressed in the rat brain and at even higher levels in rat insulinoma INS-1E cells (Fig. 1A). Co-staining of Nexmif and Ki67 in the INS-1E cells showed that Nexmif was preferentially present in duplicating INS-1E cells (Fig. 1B). We further detected Nexmif expression in whole pancreas at postnatal days 0, 7, 14, and 21 and in adult pancreatic islets (Fig. 1C), Nexmif levels decreased two weeks after birth. Similar postnatal decline has already been described in the brain3. We used mouse brain at postnatal day 3 as a positive control, as previous studies have shown a maximal expression at this time point2. Nexmif has been shown to be expressed in most tissues, although GTEx data (human) shows minimal expression in the liver. Accordingly, adult mouse liver was used as a low-expression control. Expression of Nexmif was about 10 times lower in the mouse pancreas than in the brain (Fig. 1C). However, Nexmif protein partially co-stained with insulin in islets, with very few Nexmif-positive cells in the exocrine pancreas (Fig. 1D). In human, expression of NEXMIF was about 10 times higher in pancreatic islets than in exocrine tissue (Fig. 1E). As it did in mouse pancreas, NEXMIF co-localized partially with insulin in human pancreas (Fig. 1F).

Nexmif expression in beta cells in different species and in INS-1E cells. (A) Nexmif mRNA expression in rat brain (n = 3) and in INS-1E cells (n = 3). (B) Nexmif localization (green) in proliferating INS-1E cells marked by Ki67 staining (red). DAPI staining in blue. (C) Nexmif mRNA expression from left to right in mouse brain at postnatal day 3 (MBP3) (positive control, n = 3); mouse liver at 1 year (negative control, n = 3); mouse pancreas at postnatal day 0 (n = 3), postnatal day 7 (n = 3), postnatal day 14 (n = 3), and postnatal day 21 (n = 3); and adult mouse islets (n = 3). Error bars, standard deviation; n, sample size. (D) Nexmif localization (green) in mouse pancreas at postnatal days 0 and 21, partially co-staining (yellow) with insulin (red). (E) Nexmif mRNA expression in human pancreatic endocrine (n = 3) and exocrine tissue (n = 3). (F) Nexmif localization (green) in human pancreas partially co-staining (yellow) with insulin (red). Error bars, standard deviation; n, sample size. *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
Nexmif mutant mice
To study the function of Nexmif in vivo, we used CRISPR-Cas9 technology to generate Nexmif mutant mouse lines. We obtained mice carrying a 50-nt deletion resulting in an early termination codon. Mutant mice were backcrossed with the C57BL6 strain. Nexmif protein was lost from the brain and pancreas of Nexmif mutant mice (Additional File 2: Fig. S1).
Nexmif plays a role in beta-cell proliferation
As Nexmif protein was mainly expressed in proliferating INS-1E cells, we determined if the loss of Nexmif influenced the percentage of beta cells that were proliferating in the pancreatic islets. Significantly lower percentages of proliferating beta cells were observed in the Nexmif mutant islets (Fig. 2A). Islets tended to be smaller and to contain fewer beta cells per islet in Nexmif mutant mice (Fig. 2B). This was confirmed in our islet distribution analysis, where islets containing over 200 beta cells were found only in Nexmif non-mutant mice (Fig. 2C). We measured a significantly decreased beta-cell surface (insulin positive cells, %) over total pancreas area in pancreatic sections from mutant and non-mutant Nexmif mice (P = 0.0423), suggesting a smaller total beta-cell mass (Fig. S2A). The body weight was unchanged (Fig. S2B). The percentage of beta cells undergoing apoptosis was similar in non-mutant and mutant Nexmif mice (Fig. S2C). We performed intraperitoneal glucose tolerance tests (ipGTT) at postnatal weeks 12 and 40. Randomization was done before the ipGTT by a technician so the experimenter was not aware of the genotypes of the mice. The technician revealed the group allocation for data analysis. The area under the curve was significantly higher in Nexmif mutant mice compared with control C57B6 mice outside of the colony at 40 weeks of age (Fig. 2D), while the difference was not significant versus Nexmif mutant mice of the same age. Nexmif mutant mice show however significantly increased glycemia levels at 15 min during the ipGTT in comparison to Nexmif non-mutant mice. In mutant Nexmif mice, both the insulin levels and the HOMA B%, reflecting beta-cell function, tended to be lower in comparison with the non-mutants (Fig. 2C)16.To exclude an alteration in insulin sensitivity, we calculated the quantitative insulin sensitivity check index (QUICKI)17 and found no statistical difference between the control, mutant, and non-mutant mice (P = 0.35) (Fig. 2E). To confirm the effect of Nexmif loss on beta cells in another species, we performed knockdown experiments in zebrafish. Nexmif knockdown led to a significant decrease in the beta-cell count as well as in the glucose-induced beta-cell expansion rate (Additional File 4: Fig. S3).

Pancreatic islet proliferation and function in Nexmif mutant mice. (A) Percentage of proliferating beta cells marked by Ki67 in the pancreatic islets of Nexmif non-mutant (n = 3) and mutant mice (n = 3) at postnatal day 21. Error bars indicate the standard deviation. The box plots are presented as median with 25th percentiles (boxes). Two-tailed unpaired Student t-test. ****P ≤ 0.0001. (B) Beta-cell counts in pancreatic islets of Nexmif non-mutant (n = 3) and mutant mice (n = 3) at postnatal day 21. (C) Islet size distribution in Nexmif non-mutant (n = 3) versus mutant mice (n = 3) at postnatal day 21. (D, E) Area under the curve for glucose levels and determination of insulin levels after intra-peritoneal glucose tolerance tests in control C57BL6 mice outside the colony at 12 weeks (n = 3) and 40 weeks (n = 3), Nexmif non-mutant mice at 12 weeks (n = 8) and 40 weeks (n = 13), and Nexmif mutant mice at 12 weeks (n = 12) and 40 weeks (n = 18). Only male mice were used. Error bars indicate the standard deviation and “n” is the sample size. Significance was determined using 2-way ANOVA for repeated measurements followed by a multiple comparisons post hoc test. *Control vs. Nexmif mutant, P ≤ 0.05; ***Control vs. Nexmif mutant, P ≤ 0.001; $$Nexmif non-mutant vs. Nexmif mutant, P ≤ 0.01; #Control vs. Nexmif non-mutant, P ≤ 0.05. Ns: non-significant. The quantitative insulin sensitivity check index (QUICKI) was calculated with the following formula: 1/(log(fasting insulin μU/mL) + log(fasting glucose mg/dL))17, the HOMA B% with (20 × Insulin μU/mL)/(glucose (mmol/l) – 3.5)16,66. Significance was determined using one-way ANOVA analysis. Error bars indicate the standard deviation.
Loss of Nexmif results in an increased number of DNA double-strand breaks
To determine if Nexmif could play a role in maintaining genomic stability, DNA double-strand breaks in Nexmif mutant and non-mutant pancreas were documented using the marker γ-H2AX18. There were more DNA double-strand breaks observed in Nexmif mutant than in non-mutant pancreatic islets (55% of insulin-positive cells versus 27%; Fig. 3).

DNA damage in pancreatic islets of Nexmif mutant mice. DNA double-strand breaks marked by immunofluorescent staining with γ-H2AX (green) in pancreatic islets of Nexmif non-mutant (n = 3) and mutant mice (n = 3) at postnatal day 21. Insulin (red), DAPI (blue).
Loss of Nexmif in pancreatic islets results in the downregulation of genes involved in stress response
RNAseq analysis of Nexmif mutant and control islets revealed that genes upregulated (n = 711) in the mutant islets (Fig. S4) are mainly involved in developmental processes, cell migration, or cytoskeleton organization (Gene Ontology, Biological process, Table S1). Genes that were downregulated (n = 2418) in the Nexmif mutant islets (Fig. 4B) are mainly related to stress response, protein phosphorylation and folding, regulation of molecular function, and metabolic processes (Gene Ontology, Biological process, Table S1); among them are Hsp90aa1, Hsp90ab1, H3f3b, and Zfp36 (Fig. 4B). Heat shock proteins are expressed in response to stress conditions. H3f3b encodes histone H3.3, which plays a role in beta-cell proliferation, while its main function is the maintenance of genomic integrity19,20. A big difference in expression was observed with Zfp36, which encodes a destabilizer of AU-rich element (ARE) mRNAs. Downregulation of Hsp90aa1, Hsp90ab1, H3f3b, and Zfp36 was confirmed by qRT-PCR (Fig. 4C).

RNAseq in pancreatic islets of Nexmif mutant mice. (A) Gene expression profile of differentially expressed genes in pancreatic islets of male Nexmif mutant and Nexmif non-mutant mice. Kendall’s Tau was used for clustering possibly co-expressed pathway genes (n = 3). RNAseq mRNA expression data from pancreatic islets. The most highly expressed genes that were upregulated at least twofold in Nexmif mutant islets compared with Nexmif non-mutant islets (n = 3) are shown. (B) RNAseq mRNA expression data from pancreatic islets. The most highly expressed genes that were downregulated at least twofold in Nexmif mutant islets compared with Nexmif non-mutant islets (n = 3) are shown. (C) Confirmation of downregulated genes by qPCR: Hsp90ab1, Hsp90aa1, H3f3b, Zfp36 (n = 3). Error bars indicate the standard deviation. Two-tailed unpaired Student’s t-test: *P ≤ 0.05, **P ≤ 0.01. ***P ≤ 0.001, ****P ≤ 0.0001.
Nexmif is highly expressed in response to oxidative stress
Given the downregulation of stress-response genes in the Nexmif mutant islets, we examined Nexmif expression in response to cellular stress by exposing INS-1E cells to 200 µM H2O2 over a time course. Nexmif expression was strongly increased at both the mRNA and protein levels, with maximum expression after 6 h (Fig. 5A,B). The observed cytoplasmic localization of Nexmif with a granular pattern after H2O2 treatment indicated that Nexmif could be localized in stress granules or P-bodies in the cytoplasm, in particular a few hours after the oxidative stress. Co-staining with the stress granule marker GTPase-activating protein SH3 domain–binding protein 1 (G3BP1) after H2O2 treatment suggested that Nexmif localizes partially to stress granules (Fig. 5C).

Nexmif expression in reaction to oxidative stress in INS-1E cells. (A) Nexmif mRNA expression over a time course in response to 200 µM H2O2 at time points 0 h, 0.5 h, 2 h, 6 h, and 24 h (n = 3). Error bars indicate the standard deviation. Two-tailed unpaired Student t-test: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. (B) Nexmif localization (green) in INS-1E cells challenged with 200 µM H2O2 over a time course of 0 h, 0.5 h, and 6 h by immunofluorescence. DAPI (blue). (C) Expression and co-localization of Nexmif (green) by fluorescent in situ RNA hybridization (FISH) with G3BP1 (red) in INS-1E cells, marking stress granules by immunofluorescence.
Nexmif is transcribed to form highly stable ARE mRNAs
One of the most strongly downregulated genes, Zfp36, is known as an important regulator of ARE mRNAs. ARE mRNAs are highly expressed by genes enriched with adenylate-uridylate-rich (AUUUA-elements) in their 3′ UTR. Zfp36 binds to these specific elements and plays an important role in their degradation. These elements are often found in proto-oncogenes, neuronally expressed genes such as Nexmif, and genes encoding cytokines and nuclear transcription factors21. In patients with Nexmif duplications, downregulation of gene expression has been reported22. This indicates that Nexmif might inhibit its own gene expression, potentially via the upregulation of Zfp36. It was determined that human NEXMIF contains 23 ARE sites in its 3′ UTR. Mouse Nexmif contains 15 ARE sites (AREsite, nibiru.tbi.univie.ac.at), a relatively high number. This indicates that Nexmif could be transcribed into ARE mRNAs.
To determine if Nexmif is transcribed into ARE mRNAs, Nexmif expression was determined in Elavl4 knockdown INS-1E cells. Elavl4 is a known stabilizer of ARE mRNAs23. Knockdown of Elavl4 resulted in significantly reduced expression of Nexmif in INS-1E cells (Fig. 6A). To further investigate how Nexmif is transcribed into ARE mRNAs, the localization of Nexmif mRNA was determined after cells were exposed to an oxidative stress. ARE mRNAs are known to localize to stress granules, where they are protected under stressful situations. This was indeed observed for Nexmif mRNA after oxidative stress in INS-1E cells (Fig. 6B). Additionally, to determine Nexmif mRNA stability, we performed mRNA degradation tests in INS-1E cells. Nexmif mRNA expression remained stable, while beta-actin mRNA degraded gradually when incubated at 37 °C over a time course (Fig. 6C). Actinomycin D treatment was applied to inhibit transcription. Nexmif mRNA levels increased, while beta-actin mRNA levels declined sharply within 30 min (Fig. 6D).

Determination of whether Nexmif transcripts are ARE mRNAs. (A) Elavl4 and Nexmif expression in Elavl4 knockdown INS-1E cells (n = 3). (B) Localization of Nexmif mRNA (green) and G3BP1 protein (red) after 6-h H2O2 treatment of INS-1E cells. (C) Beta-actin and Nexmif expression in INS-1E cells after incubation at 37 °C for 0 h, 0.5 h, 2 h and 6 h (n = 3). (D) Beta-actin and Nexmif expression in INS-1E cells after incubation with 10 µg/mL actinomycin D for 0 h, 0.5 h, 2 h and 6 h (n = 3). Error bars, standard deviation; n, sample size.
Loss of Nexmif induces phenotypic heterogeneity
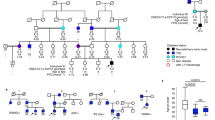
Among Nexmif mutant mice, the first founder male did not show any abnormalities. In the second generation, one heterozygous mutant female was severely growth retarded, with low-set ears and a curved spine, and died at the age of 5 weeks (Additional File 6: Fig. S5A). From the third generation onward, more abnormalities were observed. Growth-retarded pups were observed among both male and female Nexmif mutant and non-mutant mice. Whole litters were regularly born dead, and we observed 4–6-week-old juveniles having seizures resulting in death. Other but less frequent observations were hind-limb polydactyly (Additional File 6: Fig. S5B) and microphthalmia (Additional File 6: Fig. S5C).
Another interesting observation was a female mouse missing part of or a whole X-chromosome. Mutant mice were identified by a 50-nt deletion in the DNA sequence of the Nexmif gene. A heterozygous female was crossed with a wild-type C57BL6 male. As the male passes on its intact X chromosome to the female progeny, all female offspring should carry a non-mutant Nexmif allele. However, one female carried only a mutated allele. The Y-chromosomal gene Sry was not amplified by PCR, indicating that it was not a case of sex reversal. The copy number of the X-linked genes in this female was similar to that in the male control, while being half that seen in the female control (Additional File 6: Fig. S5D). Mortality rates were higher in offspring of hemizygous males carrying the Nexmif mutation than in offspring of heterozygous females carrying the Nexmif mutation. Overall, each successive generation's phenotypes appeared to be more severe (Additional File 7: Fig. S6).
Loss of Nexmif results in the activation of LINE1 elements
The expression of repeats was analyzed in the RNAseq results. In control pancreatic islets, the most abundant repeats were RNAs representing the LINE1 elements L1Md_T, L1Md_A, and L1Md_F2. In Nexmif mutant islets, expression of L1Md_T was significantly higher, while L1Md_A and L1Md_F2 showed a tendency to be upregulated (Fig. 7A). These LINE1 elements encode ORF1 protein. At postnatal day 21, ORF1 protein was highly expressed in Nexmif mutant pancreas and hardly detectable in non-mutant (Fig. 7B). As LINE1 is known to be strongly expressed in neural progenitor cells during brain development and in primordial germ cells in the testis24, we determined its expression in the brain and testis. LINE1 expression was significantly higher in the mutant brain at postnatal day 0 (Fig. 7C,D). A similar observation was made for the testis (Fig. 7E,F).

LINE1 activation. (A) RNAseq expression data for transposable LINE1 elements in pancreatic islets of 6-week-old Nexmif mutant and non-mutant mice (n = 3). (B) ORF1 protein (green) expression in Nexmif mutant and non-mutant mouse pancreas at postnatal day 21 by immunofluorescence. Insulin (red), DAPI (blue). (C) LINE1 mRNA expression in Nexmif mutant and non-mutant mouse brain at postnatal day 0 (n = 3). (D) ORF1 expression (green) in Nexmif mutant and non-mutant mouse brain (cortex) at postnatal day 0 by immunofluorescence. DAPI (blue). (E) LINE1 mRNA expression in Nexmif mutant and non-mutant mouse testis at postnatal day 0 (n = 3). ****P ≤ 0.0001. (F) ORF1 expression (green) in Nexmif mutant and non-mutant mouse testis at postnatal day 0 by immunofluorescence. DAPI (blue).
Discussion
This study started with the diagnosis of non-autoimmune diabetes and mental retardation in a 5-year old boy with a truncated NEXMIF variant. During our study, we identified a female with non-autoimmune diabetes and learning disabilities who was carrying a NEXMIF variant, underlining a role for NEXMIF in the development of diabetes. At the same time, more individuals with mental retardation and NEXMIF variants were identified25. From these patients, one additional female has been described to have diabetes, suggesting that NEXMIF plays a role in diabetes development. We hypothesize that an inadequate beta-cell mass prompted diabetes in both patients, as insufficient beta-cell mass is key in the pathogenesis of diabetes. Indeed, we demonstrated both RNA and protein NEXMIF/Nexmif expression in pancreatic beta cells. The nuclear and cytoplasmic localization we observed in beta cells has been described in brain neurons by Ishikawa et al.3. The metabolic microenvironment could influence the localization of the protein, as has been observed of the beta-cell transcription factor Pdx126,27.
The RNAseq data show decreased expression of H3f3b, encoding H3.3. Proper deposition of histone H3.3 has been shown to play a role in human beta-cell proliferation and could be a cause of reduced beta-cell proliferation in Nexmif mutant islets19.
One of the most strongly downregulated gene in the mutant islets was Zfp36. As Zfp36 is an ARE-binding factor, we tested whether Zpf36 might affect Nexmif itself, as Nexmif could potentially repress its own expression22 through the upregulation of Zfp36. The presence of a significant number of ARE sites in the Nexmif 3′ UTR, plus the fact that Nexmif localizes to stress granules after oxidative stress, makes this hypothesis plausible. RNA-binding proteins and mRNA processing are emerging mechanisms regulating beta-cell function and survival.
One important observation is that the patients described in the literature have a highly variable phenotype. They are all mentally retarded, but the associated phenotype differs, even among family members with the same mutation. Additionally, females carrying NEXMIF mutations can be either severely mentally retarded or unaffected. This does not seem to be due to X-inactivation, which has been shown to be random in both patients and unaffected carriers. Unaffected female carriers are known to inherit NEXMIF mutations maternally, while affected females acquire it de novo, indicating a parent-of-origin effect. Previous work on the silencing of Nexmif in the developing mouse brain indicated that Nexmif plays a role in neuronal migration and dendritic growth28. Increased cell surface expression of N-cadherin as well as altered delta-catenin signaling and actin dynamics explain the impaired neuronal morphogenesis at the molecular level. In addition, knockdown experiments in neuronal cells have also shown upregulation of beta-integrin, which is implicated in enhanced cell–cell adhesion29.
We studied a mouse model carrying a truncated Nexmif variant. Interestingly, we observed high mortality and high phenotypic variability between siblings, independent of the Nexmif mutation. The mortality rate and phenotypic variability seemed to increase with each generation, and even more severely when the carrier parent was a male rather than a female. This supports the notion of a parent-of-origin effect, as indicated by observations in NEXMIF families. Other phenotypes shared between our mouse model and NEXMIF patients are growth retardation, obesity, and seizures.
Our study indicates that the observed variability could be due to a loss of genomic integrity. We observed an increase in DNA double-strand breaks in the pancreas of Nexmif mutant mice. Additionally, RNAseq on pancreatic islets indicated a downregulation of genes involved in the stress response. Interestingly, Hsp90, which was found to be downregulated in Nexmif mutant islets, plays an important role in neurite outgrowth and stress response. Most importantly, Hsp90 has also been identified as an evolutionary capacitor, allowing a population to accrue genetic variation when insufficiently expressed. Lack of Hsp90 expression results in phenotypic variation in the offspring of Drosophila and cave fish, including offspring that re-express Hsp9030,31. This is interesting, as we observed high variation in our mouse Nexmif colony, including in offspring carrying the non-mutant Nexmif allele. Several studies have reported that the activation of transposable elements plays a role in causing variation. In flies and mammals, Hsp90 has been shown to bind chromatin at gene regulatory elements, where it stabilizes transcriptional and epigenetic factors32,33,34,35. Additionally, in Drosophila a lack of Hsp90 results in the transposition of transposable elements (TEs), possibly disrupting important genes. In Drosophila and mice, Hsp90 has been shown to control the transposition of transposable LINE1 elements in the male germ line via piRNA pathways36,37. Another downregulated gene that is known to play an important role in maintaining genomic integrity is H3f3b. H3f3b and H3f3a both encode the highly conserved protein, histone H3.3. Lack of histone H3.3 induces dysfunction of heterochromatin structures at pericentric regions and telomeres, resulting in mitotic and meiotic defects38,39,40. It is likely that H3f3b (H3.3) regulates LINE1 elements, as H3.3 deposited in heterochromatic genomic regions over LINE1 elements represses LINE119,20,41. A decrease in H3.3, as shown here in Nexmif mutant mice, leads to an increase in LINE1 element expression. The observation of a female missing a part or all of one X chromosome suggests that the loss of Nexmif can result in meiotic defects, which could be due to the downregulation of H3f3b. Histone H3.3 has also been shown in embryonic stem cells to be essential for silencing transposons, among which LINE1 elements cause a loss of genomic integrity41. Additionally, histone 3.3 has been shown to be essential for proper neuronal differentiation, neurite outgrowth, and proliferation42. Histone H3.3 deposition occurs during development and also after UV-induced DNA damage43,44. The downregulation of Hf3bf in Nexmif mutant mice as well as the importance of histone H3.3 deposition during brain development and DNA damage response indicate that there might be an important link between histone H3.3 and Nexmif. As previously mentioned, Nexmif was suggested to have a similar function as Atrx3. Interestingly, Atrx has been shown to bind and control histone H3.3 deposition at pericentric regions, telomeres, and silenced methylated regions to maintain epigenetic modifications such as H3K9me339,45. Another parallel with Atrx is the observation of polydactyly in one mouse of a colony lacking Atrx expression in chondrocytes. Polydactyly did not seem to be a direct effect of the loss of Atrx, as other mice in the colony were not affected46. Polydactyly is rare and the mouse we identified in our colony was not a carrier of the Nexmif mutation, indicating an indirect effect. Loss of Nexmif expression might not only regulate histone H3.3 expression but also H3.3 deposition44,46. A possible physical association of Nexmif with histone H3.3 should be determined in future studies.
Both Hsp90 and histone H3.3 have been implicated in LINE1 inhibition41. We observed increased LINE1 expression in the pancreas, brain, and testis of Nexmif mutant mice. LINE1 activity could be detrimental to genomic integrity, as the LINE1 mRNA encodes two proteins (ORF1 and ORF2) with endonuclease and retro-transcriptase activity. These two proteins bind to the LINE1 mRNA, create double-strand breaks in DNA, and aid in the retrotransposition of LINE1 back into the genome. LINE1 elements play a major role in evolution and variability in a population due to their ability to retrotranspose in the germ line47,48,49,50,51. Additionally, they have been shown to play a major role in regulating chromatin structure and accessibility during development and to regulate the expression of many other genes52. LINE1 elements have been shown to be highly active in neural progenitors, but also in the male germ line. Increased LINE1 activity is associated with oxidative stress, autism, schizophrenia, Alzheimer’s disease, auto-immune diseases, ataxia telangiectasia and Rett syndrome53,54,55,56,57,58. Rett syndrome patients have characteristics similar to those of NEXMIF patients. The mutated gene in Rett syndrome, MECP2, is also involved neurite outgrowth and LINE1 inhibition54. These observations and our results could imply a similar role for NEXMIF.
Methods
Genetic analysis
Exomic regions of patient and parent DNA were selected with liquid phase capture (Sureselect XT, Agilent, Santa Clara, CA, USA) and analyzed with massive parallel sequencing in an Illumina HiSeq2000 high-throughput sequencer, using the manufacturer's protocols. We considered only exonic and splicing variants (± 10 bp from the intron–exon boundary) for further study. We excluded synonymous variants with a minimum allele frequency ≥ 0.01 in the dbSNP database. Remaining variants were checked in the Exome Variant Server, ExAC, and gnomAD databases. The pathogenic NEXMIF variant was confirmed with Sanger sequencing.
We excluded all variants inherited from either parent in the trio under the hypothesis of de novo mutations as the underlying cause of the syndromic diabetes. The variant was confirmed by Sanger sequencing. The absence of NEXMIF-truncating variants in the gnomAD (https://gnomad.broadinstitute.org/) is consistent with a genetic disease due to NEXMIF loss of function.
Raw sequencing data were analyzed using a locally developed pipeline integrating BWA, SAM, PINDEL, and ANNOVAR algorithms for alignment on the hg19 human reference genome and the identification and annotation of variants, including the interrogation of databases for known polymorphisms and mutations59. The most prominent variants were selected according to the prediction score for damaging effects on the respective proteins using SIFT (score < 0.05), Polyphen-2 (score > 0.85), MutationTaster-2 (score > 0.5), the likelihood-ratio test LRT (score > 0.9), PhyloP (score > 0.85), and GERP (score > 2) and confirmed by Sanger sequencing. Variants with an allelic frequency ≥ 0.1% in the 1000 Genomes Project, Exome Variant Server database were filtered out.
Cell culture
Clonal INS-1E cells (RRID:CVCL_0351, obtained from P. Maechler) were plated at a concentration of 2 × 105 cells/mL in a 12-well plate (1 mL/well) and cultured for 72 h in a humidified atmosphere containing 5% CO2 in complete medium composed of GlutaMax RPMI-1640 (ThermoFisher Scientific 61870036) supplemented with 5% heat-inactivated fetal calf serum (Gibco 10500064), 1 mM sodium pyruvate, 50 μM β-mercaptoethanol (Gibco 21985023), 10 mM HEPES, 100 μg/mL streptomycin, and 100 U/mL penicillin60.
Cell fixing and staining
Cells were cultured as described, in wells containing coverslips coated with poly-L-ornithine (Sigma, P4957). To fix the cells, the medium was aspirated and the coverslips were covered with 4% PFA for 6 min. Coverslips were washed three times with PBS and covered with a 3% BSA in PBS solution for 1 h. Primary antibodies were diluted in 3% BSA and slides were incubated at 4 °C overnight. Coverslips were washed 3 times with PBS and secondary antibodies in PBS (1:500) were added for 1 h at room temperature. Coverslips were washed 3 times with PBS and covered in DAPI in PBS (1:50,000, Invitrogen D1306) for 5 min. Coverslips were then washed 3 times with PBS. Mounting medium was applied to microscope slides and the inverted coverslips were placed on top. Slides were kept at 4 °C. The primary and secondary antibodies used and their dilutions are presented in Additional File 9: Table S2.
Quantitative real-time PCR
RNA extraction was performed with a Qiagen RNAeasy mini kit (Qiagen 74136) using the manufacturer’s instructions. RNA (3 µg, 0.1 µg/µL) was reverse transcribed to cDNA using 0.5 µg random hexamers (Microsynth), 400 U Moloney murine leukemia virus reverse transcriptase (Invitrogen, 28025-013), 20 U recombinant RNasin (Promega, N251B), 2 mmol/L dNTPs (Rovalab, R203), and 40 mmol/L dithiothreitol (Invitrogen). Quantitative real-time PCR was performed with the Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659) and using an ABI StepOne Plus Sequence Detection System (Applera Europe). Gene expression data were normalized using the housekeeping gene 18S RNA unless otherwise stated. qPCR primers used in this study can be found in Additional File 10: Table S3.
cDNA from INS-1E cells after knockdown of Elavl4 by siRNA was kindly provided by the laboratory of Prof D. Eizirik, ULB Center for Diabetes Research, Université Libre de Bruxelles, Belgium61.
Tissue collection
Brain, pancreas, and testis were collected at postnatal days 0, 3, 7, 14, and 21 and/or 42. Tissue was snap frozen before RNA extraction or immersed in a 4% PFA solution overnight. Tissue was then transferred to PBS for 24 h and afterwards dehydrated and embedded in paraffin, and 5-μm sections were prepared. For analysis of proliferative cells, 5 sections at 50-μm intervals were selected from each pancreas.
Immunofluorescent staining of tissue sections
Tissue slides were twice covered in Neoclear solution for 5 min. To rehydrate, slides were immersed in the following solutions: 100% ethanol, 100% ethanol, 95% ethanol, 70% ethanol, 50% ethanol, and 35% ethanol. Slides were washed three times in PBS. Next, slides were heated for 20 min in citrate buffer and washed three times in PBS. Slides were permeabilized with 0.3% Triton X-100 in PBS for 10 min. Blocking was performed with 2% BSA, 0.3% Triton X-100, and 5% goat serum for 30 min. Slides were incubated with 2% BSA, 1% goat serum, and the primary antibody of interest in PBS solution overnight. Slides were washed three times with PBS and incubated with the corresponding secondary antibody in PBS (1:1000) for 2 h at RT. Slides were then washed three times and incubated with DAPI in PBS (1:50,000). Next, slides were washed three times with PBS and mounted. Slides were analyzed by use of a Nikon A1r spectral confocal microscope. Slides used for determining the number of proliferating cells were scanned with a Zeiss Axio Scan.Z1 wide-field scanner. Antibodies used in this study are presented in Additional File 9: Table S2.
H2O2 treatment INS-1E cells
INS-1E cells were cultured as described above. After culturing for 72 h, cells were treated with 200 µM H2O2 in order to induce a stress response62. Cells were collected after 30 min, 2 h, 6 h, and 24 h for RNA extraction and immunofluorescent staining.
Generation of Nexmif mutant mice
We consulted different web resources to design the following top- and bottom-strand SgRNA oligos to target Nexmif specifically63, https://zlab.bio/guide-design-resources):
Sgrna1
-
5′ caccgAATGATCGGGTGCTTCAATC
-
cTTACTAGCCCACGAAGTTAGcaaa 5′ ⇒ 5′
-
aaacGATTGAAGCACCCGATCATTc.
Sgrna2
-
5′ caccgCACGGCTTCTTAGATGGCGG
-
cGTGCCGAAGAATCTACCGCCcaaa 5′ ⇒ 5′
-
aaacCCGCCATCTAAGAAGCCGTGc
2 µg of top- and bottom-strand oligos was added to a total volume of 50 µL Neb2 buffer. Samples were heated for 3 min at 95 °C and cooled down for 45 min at RT. Next, 5 µL of annealed oligos were diluted with 45 µL nuclease-free water and the concentration was determined by Nanodrop. Next, 5 µg of PX330 was digested with Bbsl in a total volume of 50 µL for 3 h at 37 °C. The digested PX330 was loaded on a 0.5% agarose gel and excised and purified with a gel extraction kit (Qiagen QIAquick, Qiagen 28,106, manufacturer’s protocol). A sample was run on a 2% agarose gel to verify the presence of purified PX330. Next, 1 µL annealed oligos (75–600 ng), 1.5 µL PX330 Bbsl vector (± 100 ng), 1 µL T4 ligase buffer, 1 µL T4 ligase enzyme, and 5.5 µL nuclease-free water were mixed. Samples were incubated at RT for 10 min and then at 65 °C for 10 min. Ligated PX330-SgRNA was used to transform Max Efficiency DH5α Competent Cells using the manufacturer's protocol (Invitrogen 18265017). We screened for the presence of PX330 by PCR, using PX330 primer 5′ GGCCTATTTCCCATGATTCC 3′ and the bottom-strand SgRNA oligo. Colonies were picked, put in the PCR mix, and incubated overnight in mini-culture at 37 °C. The PCR product was run on a 2% agarose gel, following the manufacturer’s instructions for the mini-preparation (Qiagen QIAprep Spin miniprep). PX330 plasmids containing SgRNA1 and SgRNA2 were selected and injected into DBA2 oocytes fertilized with C57BL6 sperm. We generated a genetically modified mouse line from a male with a truncating Nexmif variant p.Pro68* due to a 50-nucleotide deletion, as it would most likely be the most detrimental. The top 3 most likely off-target regions (https://crispr.cos.uni-heidelberg.de/) were confirmed by PCR and Sanger sequencing to be not affected. Mice were backcrossed with C57BL6 mice for at least 5 generations before being used in experiments. We used a total of 118 animals. The sample size was chosen according to standards in the field.
Genotyping
Ear punches were collected and immersed in lysis buffer (Viagen Biotech 102-t) with proteinase K (Macherey Nagel 740506). Samples were incubated at 55 °C for 8 h and then at 85 °C for 1 h. Screening was performed using Nexmif primers: Primer F: TCTACCTTTTCCTGGGCTGA, Primer R: AGCAACCTAAGCAAGTCCTG. Accuprime (Life Technologies 12339024, manufacturer’s protocol) was used for the DNA amplification. The resulting product was 600 bp in length.
Glucose intolerance measurements
Glucose (2 g/kg body weight) was administered intra-peritoneally in 6-h-fasted male mice before measurements of glucose levels. Glucose measurements (Accu-Check, Roche Diagnostics, Rotkreuz, Switzerland) and blood collection (Sarstedt low-bind microtubes) from the tail vein were performed at times 0 min, 15 min, and 30 min. Glucose levels were additionally measured at 60 min and 90 min.
Insulin level measurements
Plasma insulin levels from blood sampled at 0 min, 15 min, and 30 min after glucose administration were determined using an ultrasensitive mouse insulin ELISA (Mercodia AB, Uppsala, Sweden).
Islet isolation
Pancreatic islets were isolated from 6-week-old male mice, using collagenase P (Roche 11249002001) with a Histopaque (Sigma-Aldrich 10771) gradient as described previously64. After the gradient step, RNA was extracted from hand-picked islets.
RNAseq
Gene expression in pancreatic islets from 6-week-old male Nexmif mutant and non-mutant mice was profiled with the Affymetrix Genechip system. Sequencing quality control was done with FastQC v.0.11.5. Reads were mapped with the TopHat v.0.2.1.1 software to the USCS mm10 reference genome. The table of counts with the number of reads mapping to each gene feature on UCSC mm10 was prepared with HTSeq v0.6p1. The differential expression analysis was performed with the statistical analysis R/Bioconductor package edgeR v. 3.14.0. Briefly, the counts were normalized according to the library size and filtered65. The genes having a count above 1 count per million reads (cpm) in at least 3 samples were kept for the analysis. Additionally, repeats were analyzed. Selected differentially expressed genes were verified by qPCR (at least twofold difference, with an SD < 25% of the average value).
In situ hybridization
Samples were fixed with 4% paraformaldehyde for 6 min and then permeabilized in 0.3% Triton X-100 in PBS. After three rinses with PBS, the samples were rinsed 2 times with 1 × saline sodium citrate (SSC)/50% formamide, fluorescent probes were added, and FISH was performed overnight at 37 °C (5 µL of probes in 500 µL solution). FISH probes were DNA oligos labeled with a ULYSIS Alexa Fluor 546 Nucleic Acid Labeling Kit (following the manufacturer’s protocol).
Slides were washed three times with PBS and incubated for 30 min with 5% BSA PBS solution for blocking. Next, the slides were incubated for 2 h with G3BP1 antibody (1:200) in PBS, after which they were washed three times with PBS. The slides were then incubated for 1 h with secondary antibody (donkey anti-goat Alexa Fluor 488), washed three times with PBS, and incubated with DAPI for 5 min. Finally, the slides were washed three times with PBS and coverslipped with mounting medium.
Actinomycin D treatment
INS-1E cells were cultured as described above. After being cultured for 72 h, cells were treated with 10 nM actinomycin D (Sigma, A1410). Cells were collected after 30 min, 2 h, and 6 h for RNA extraction.
mRNA degradation
INS-1E cells were cultured as described above. After being cultured for 72 h, cells were collected and mRNA was extracted in a total volume of 45 µL. The extracted mRNA was redistributed in 10-µL aliquots to 4 Eppendorf tubes. The tubes were incubated at 37 °C for 0 h, 0.5 h, 2 h, and 6 h. RT-PCR was performed and the degradation of Actb (beta actin) and Nexmif mRNAs was determined by the Ct value.
Zebrafish methods
See Additional File 1.
Statistical analysis
Paired t-test analyses were performed with Prism GraphPad software (v.8.4.2) when appropriate. Data are presented as means ± standard error of the mean (SEM) and are significant at *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
Ethics approval and consent to participate
We obtained informed consent to participate from all the participants or their legal guardians. The study was approved by the “Commission Centrale D’éthique de la Recherche sur L’être Humain” (#CER 11-140-2°).
All animal-based research was conducted at the University of Geneva Medical Center with the approval of the animal care and experimentation authorities of the Canton of Geneva (#GE/153/16).
We confirm that all methods were performed in accordance with the relevant guidelines and regulations.
We also confirm that the study is reported in accordance with ARRIVE guidelines.
Consent for publication
We obtained consent for publication from the participants or their legal guardians.
Web resources
GTExportal, (https://gtexportal.org/home/gene/KIAA2022, SIFT, http://sift.jcvi.org, PolyPhen2, http://genetics.bwh.harvard.edu/pph2/, Mutation Taster, http://www.mutationtaster.org, Likelihood-ratio test query, http://www.genetics.wustl.edu/jflab/lrt_query.html, GnomAD, https://gnomad.broadinstitute.org/, Zhang lab, https://zlab.bio/guide-design-resources, CCTop, https://crispr.cos.uni-heidelberg.de/.
Data availability
The data generated or analyzed during this study are included in this article and its supplemental information files or will be available from the following repository: https://zenodo.org/.
References
Stamberger, H. et al. NEXMIF encephalopathy: An X-linked disorder with male and female phenotypic patterns. Genet. Med. 23, 363–373 (2021).
Cantagrel, V. et al. Spatiotemporal expression in mouse brain of Kiaa 2022, a gene disrupted in two patients with severe mental retardation. Gene Express. Patterns. 9, 423–429 (2009).
Ishikawa, T. et al. Transient expression of Xpn, an XLMR protein related to neurite extension, during brain development and participation in neurite outgrowth. Neuroscience 214, 181–191 (2012).
Martin, S. K. & Wood, R. D. DNA polymerase ζ in DNA replication and repair. Nucleic Acids Res. 47, 8348–8361 (2019).
Lin, W., Wu, X. & Wang, Z. A full-length cDNA of hREV3 is predicted to encode DNA polymerase zeta for damage-induced mutagenesis in humans. Mutat. Res. 433, 89–98 (1999).
Sloun, P. P. H. V. et al. Involvement of mouse Rev3 in tolerance of endogenous and exogenous DNA damage. Mol. Cell. Biol. 22, 2159–2169 (2002).
Tomas-Roca, L. et al. De novo mutations in PLXND1 and REV3L cause Möbius syndrome. Nat. Commun. 6, 7199 (2015).
Gilbert, J. et al. NEXMIF/KIDLIA knock-out mouse demonstrates autism-like behaviors, memory deficits, and impairments in synapse formation and function. J. Neurosci. 40, 237–254 (2020).
Rubio-Cabezas, O. et al. Homozygous mutations in NEUROD1 are responsible for a novel syndrome of permanent neonatal diabetes and neurological abnormalities. Diabetes 59, 2326–2331 (2010).
Stekelenburg, C. M. & Schwitzgebel, V. M. Genetic defects of the β-cell that cause diabetes. Endocr. Dev. 31, 179–202 (2016).
Shaw-Smith, C. et al. GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes 63, 2888–2894 (2014).
Gloyn, A. L. et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N. Engl. J. Med. 350, 1838–1849 (2004).
Bonnefond, A., Philippe, J., Durand, E. & Dechaume, A. Whole-exome sequencing and high throughput genotyping identified KCNJ11 as the thirteenth MODY gene. PLoS ONE 7, e37423 (2012).
He, Q. et al. The Daxx/Atrx complex protects tandem repetitive elements during DNA hypomethylation by promoting H3K9 trimethylation. Stem Cell. 17, 273–286 (2015).
Lambert, N. et al. Novel NEXMIF pathogenic variant in a boy with severe autistic features, intellectual disability, and epilepsy, and his mildly affected mother. J. Hum. Genet. 63, 847–850 (2018).
Maugham, M. L. et al. Insights from engraftable immunodeficient mouse models of hyperinsulinaemia. Sci. Rep. 7, 491 (2017).
Berglund, E. D. et al. Glucose metabolism in vivo in four commonly used inbred mouse strains. Diabetes 57, 1790–1799 (2008).
Turinetto, V. & Giachino, C. Multiple facets of histone variant H2AX: A DNA double-strand-break marker with several biological functions. Nucleic Acids Res. 43, 2489–2498 (2015).
Paul, P. K. et al. Histone chaperone ASF1B promotes human β-cell proliferation via recruitment of histone H3.3. Cell Cycle 15, 3191–3202 (2016).
Jang, C.-W., Shibata, Y., Starmer, J., Yee, D. & Magnuson, T. Histone H33 maintains genome integrity during mammalian development. Genes Dev. 29, 1377–1392 (2015).
Chen, C. Y. & Shyu, A. B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 20, 465–470 (1995).
Charzewska, A. et al. A duplication of the whole KIAA2022gene validates the gene role in the pathogenesis of intellectual disability and autism. Clin. Genet. 88, 297–299 (2014).
Bronicki, L. M. & Jasmin, B. J. Emerging complexity of the HuD/ELAVl4 gene: Implications for neuronal development, function, and dysfunction. RNA 19, 1019–1037 (2013).
Coufal, N. G. et al. L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131 (2009).
Collaborative, E. Ultra-rare genetic variation in the epilepsies: A whole-exome sequencing study of 17,606 individuals. Am. J. Hum. Genet. 105, 267–282 (2019).
Hagman, D. K., Hays, L. B., Parazzoli, S. D. & Poitout, V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans*. J. Biol. Chem. 280, 32413–32418 (2005).
Macfarlane, W. M. et al. Glucose stimulates translocation of the homeodomain transcription factor PDX1 from the cytoplasm to the nucleus in pancreatic β-cells*. J. Biol. Chem. 274, 1011–1016 (1999).
Gilbert, J. & Man, H. Y. The X-linked autism protein KIAA2022/KIDLIA regulates neurite outgrowth via N-cadherin and -catenin signaling. ENeuro. 3, 1–17 (2016).
Magome, T. et al. XLMR protein related to neurite extension (Xpn/KIAA2022) regulates cell-cell and cell-matrix adhesion and migration. Neurochem. Int. 63, 561–569 (2013).
Specchia, V. et al. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 463, 662–665 (2010).
Hummel, B. et al. The evolutionary capacitor HSP90 buffers the regulatory effects of mammalian endogenous retroviruses. Nat. Struct. Mol. Biol. 24, 234–242 (2017).
Sawarkar, R., Sievers, C. & Paro, R. Hsp90 globally targets paused RNA polymerase to regulate gene expression in response to environmental stimuli. Cell 149, 807–818 (2012).
Greer, C. B. et al. Histone deacetylases positively regulate transcription through the elongation machinery. CELREP. 13, 1444–1455 (2015).
Sawarkar, R. & Paro, R. Hsp90@chromatin.nucleus: An emerging hub of a networker. Trends Cell Biol. 23, 193–201 (2013).
Sollars, V. et al. Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat. Genet. 33, 70–74 (2003).
Gangaraju, V. K. et al. Drosophila Piwi functions in Hsp90-mediated suppression of phenotypic variation. Nat. Genet. 43, 153–158 (2011).
Ichiyanagi, T. et al. HSP90α plays an important role in piRNA biogenesis and retrotransposon repression in mouse. Nucleic Acids Res. 42, 11903–11911 (2014).
Yuen, B. T. K. & Knoepfler, P. S. Histone H3.3 mutations: A variant path to cancer. Cancer Cell 24, 567–574 (2013).
Lewis, P. W., Elsaesser, S. J., Noh, K.-M., Stadler, S. C. & Allis, C. D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 107, 14075–14080 (2010).
Sakai, A., Schwartz, B. E., Goldstein, S. & Ahmad, K. Transcriptional and developmental functions of the H3.3 histone variant in drosophila. Curr. Biol. 19, 1816–1820 (2009).
Elsässer, S. J., Noh, K.-M., Diaz, N., Allis, C. D. & Banaszynski, L. A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240–244 (2015).
Xia, W. & Jiao, J. Histone variant H3.3 orchestrates neural stem cell differentiation in the developing brain. Cell Death Diff. 24, 1548–1563 (2017).
Kim, H., Heo, K., Choi, J., Kim, K. & An, W. Histone variant H33 stimulates HSP70 transcription through cooperation with HP1γ. Nucleic Acids Res. 39, 8329–8341 (2011).
Adam, S., Polo, S. E. & Almouzni, G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell 155, 94–106 (2013).
Voon, H. P. J. & Wong, L. H. New players in heterochromatin silencing: Histone variant H3.3 and the ATRX/DAXX chaperone. Nucleic Acids Res. 44, 1496–1501 (2016).
Solomon, L. A., Li, J. R., Bérubé, N. G. & Beier, F. Loss of ATRX in chondrocytes has minimal effects on skeletal development. PLoS ONE 4, e7106 (2009).
Huang, C. R. L., Burns, K. H. & Boeke, J. D. Active transposition in genomes. Annu. Rev. Genet. 46, 651–675 (2012).
Wang, P. J. Tracking LINE1 retrotransposition in the germline. Proc. Natl. Acad. Sci. USA. 114, 7194–7196 (2017).
Xing, J., Witherspoon, D. J., Ray, D. A., Batzer, M. A. & Jorde, L. B. Mobile DNA elements in primate and human evolution. Am. J. Phys. Anthropol. 134, 2–19 (2007).
Faulkner, G. J. & Garcia-Perez, J. L. L1 mosaicism in mammals: Extent, effects, and evolution. Trends Genet. 33, 802–816 (2017).
Boissinot, S., Chevret, P. & Furano, A. V. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Mol. Biol. Evol. 17, 915–928 (2000).
Jachowicz, J. W. et al. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat. Genet. 49, 1502–1510 (2017).
Bundo, M. et al. Increased L1 retrotransposition in the neuronal genome in schizophrenia. Neuron 81, 306–313 (2014).
Muotri, A. R. et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446 (2010).
Coufal, N. G. et al. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc. Natl. Acad. Sci. USA 108, 20382–20387 (2011).
Shpyleva, S., Melnyk, S., Pavliv, O., Pogribny, I. & James, S. J. Overexpression of LINE-1 retrotransposons in autism brain. Mol. Neurobiol. 55, 1740–1749 (2018).
Mavragani, C. P. et al. Expression of long interspersed nuclear element 1 retroelements and induction of type i interferon in patients with systemic autoimmune disease. Arthritis Rheumatol. 68, 2686–2696 (2016).
Guo, S. et al. GATA4 as a novel regulator involved in the development of the neural crest and craniofacial skeleton via Barx1. Cell Death Diff. 25, 1996–2009 (2018).
Santoni, F. A. et al. Simultaneous identification and prioritization of variants in familial, de novo, and somatic genetic disorders with VariantMaster. Genome Res. 24, 349–355 (2014).
Merglen, A. et al. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 145, 667–678 (2004).
Juan-Mateu, J. et al. Neuron-enriched RNA-binding proteins regulate pancreatic beta cell function and survival. J. Biol. Chem. 292, 3466–3480 (2017).
Li, N. et al. Transient oxidative stress damages mitochondrial machinery inducing persistent beta-cell dysfunction. J. Biol. Chem. 284, 23602–23612 (2009).
Montague, T. G., Cruz, J. M., Gagnon, J. A., Church, G. M. & Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 42, W401–W407 (2014).
Carobbio, S. et al. Deletion of glutamate dehydrogenase in beta-cells abolishes part of the insulin secretory response not required for glucose homeostasis. J. Biol. Chem. 284, 921–929 (2009).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25–R29 (2010).
Wallace, T. M., Levy, J. C. & Matthews, D. R. Use and abuse of HOMA modeling. Diabetes Care 27, 1487–1495 (2004).
Acknowledgements
We thank Taina Carreaux and Fabrizio Thorel for helping to create the knockout mice and Jérémy Bévillard for technical assistance. We thank Decio Eizirik for sending us the INS-1E cells with the Huv knockdown.
Funding
The study was supported by a grant from the Swiss National Science Foundation Grant No. CR33I3_140655 and CR33I3_166591 to Schwitzgebel VM.
Author information
Authors and Affiliations
Contributions
V.S. conceived the project and acquired the main funding. The genetic analyses were performed and analyzed by J.L.B. and F.S. C.S. and R.D. performed the animal and cell experiments. C.S. wrote the manuscript with inputs from co-authors. V.S., P.M., J.L.B., and R.D. reviewed and revised the manuscript. The studies in zebrafish were designed and carried out by N.Z. and E.O.H.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stekelenburg, C., Blouin, JL., Santoni, F. et al. Loss of Nexmif results in the expression of phenotypic variability and loss of genomic integrity. Sci Rep 12, 13815 (2022). https://doi.org/10.1038/s41598-022-17845-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-17845-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
