
Abstract
Vascular endothelial (VE)-cadherin in endothelial adherens junctions is an essential component of the vascular barrier, critical for tissue homeostasis and implicated in diseases such as cancer and retinopathies. Inhibitors of Src cytoplasmic tyrosine kinase have been applied to suppress VE-cadherin tyrosine phosphorylation and prevent excessive leakage, edema and high interstitial pressure. Here we show that the Src-related Yes tyrosine kinase, rather than Src, is localized at endothelial cell (EC) junctions where it becomes activated in a flow-dependent manner. EC-specific Yes1 deletion suppresses VE-cadherin phosphorylation and arrests VE-cadherin at EC junctions. This is accompanied by loss of EC collective migration and exaggerated agonist-induced macromolecular leakage. Overexpression of Yes1 causes ectopic VE-cadherin phosphorylation, while vascular leakage is unaffected. In contrast, in EC-specific Src deficiency, VE-cadherin internalization is maintained and leakage is suppressed. In conclusion, Yes-mediated phosphorylation regulates constitutive VE-cadherin turnover, thereby maintaining endothelial junction plasticity and vascular integrity.
Similar content being viewed by others

Main
Endothelial cell–cell adhesions form an integral part of the vascular barrier, restricting the passage of molecules and cells across the vessel wall1. The barrier in most organs is flexible, allowing remodeling of the embryonic and postnatal vascular beds and exchange of small molecules and fluid to maintain adult tissue homeostasis2. During acute inflammation and in a range of chronic diseases, the barrier is weakened, leading to extravasation of blood constituents, promoting inflammation and progression of pathologies such as retinopathies and cancer1,2,3. The maintenance of junctions and barrier function relies on junctional complexes consisting of cell-adhesion molecules formed between adjacent ECs. Tight junctions limit vascular leakage preferentially in the central nervous system (CNS) and in arteries and post-arterial capillaries in peripheral tissues4,5,6. Adherens junctions, formed by VE-cadherin and associated catenins, are crucial for regulating venous and capillary vascular permeability, leukocyte extravasation and collective cell migration7,8,9,10,11,12,13. VE-cadherin is rapidly internalized in response to inflammatory cytokines and vascular endothelial growth factor A (VEGFA), allowing transient opening of gaps at EC contacts1. Although tyrosine phosphorylation of VE-cadherin has been implicated in the regulation of its internalization, the mechanisms of gap formation and vascular leakage have remained poorly understood14,15. This is important, as exaggerated vascular leakage and the formation of edema are drivers of disease1.
Three main tyrosine phosphorylation sites have been identified in VE-cadherin, Y658, Y685 and Y731 (refs. 16,17). Both Y658 and Y685 are phosphorylated in veins, but not in arteries, as a consequence of flow-induced activation of Src cytoplasmic tyrosine kinases18. Phosphorylated (p)Y658 relieves tension across VE-cadherin bridges through binding of the polarity protein LGN (leucine–glycine–asparagine repeat), which displaces VE-cadherin-associated p120-catenin19. Phosphorylation of Y685 is induced by flow as well as by inflammatory cytokines and VEGFA17,18,20. Vascular leakage requires phosphorylation of Y685, as inferred from a Y685-to-phenylalanine (F) exchange mutant mouse, which shows suppressed extravasation of large molecular weight tracers or cells in agonist-treated healthy tissues and in disease models11,21,22. Leakage is accompanied by VE-cadherin endocytosis and reduced levels of pVE-cadherin18. Y731 is constitutively phosphorylated and its dephosphorylation by Src homology phosphatase 2 (SHP2) is a prerequisite for leukocyte extravasation11,23. Several additional phosphotyrosine phosphatases (PTPs), such as VE-PTP and density enhanced phosphatase 1 (DEP1; also denoted CD148), have been implicated in the regulation of VE-cadherin phosphorylation24,25,26.
Src-mediated VE-cadherin phosphorylation on Y685 in response to VEGFA stimulation has been regarded as an important step in VEGFA-induced leakage11,17,19; however, EC-specific Src deficiency interferes with cell-matrix adhesion rather than cell–cell adhesion27,28. In addition to Src, ECs express the highly related cytoplasmic tyrosine kinases Yes and Fyn, the roles of which have remained poorly understood. In vitro studies have suggested distinct roles for Src, Yes and Fyn in regulating EC behaviors, despite their well-conserved Src homology 2 (SH2), SH3 and kinase domains29. Accordingly, constitutive global gene inactivation results in abolished VEGFA-induced vascular leakage in constitutive gene inactivated Src−/− or Yes1−/− mice but not in Fyn−/− mice30.
Here, we demonstrate a critical role for Yes in the phosphorylation of VE-cadherin at all main phosphorylation sites, Y658, Y685 and Y731. Loss of VE-cadherin phosphorylation suppresses its endocytosis and interferes with cell–cell adhesion, leading to abnormal barrier properties with blunted collective migration, excessive macromolecular leakage and suppressed leukocyte extravasation. Forced phosphorylation of VE-cadherin by Yes1 overexpression does not affect agonist-induced leakage; thus, macromolecular leakage is not controlled solely by the extent of VE-cadherin phosphorylation. In contrast, in Src deficiency, VE-cadherin endocytosis is unaffected and vascular leakage is reduced. Therefore, Yes and Src differentially regulate VE-cadherin turnover, adherens junction stability and vascular leakage. We conclude that the two highly related cytoplasmic kinases have distinct and even opposing roles in the vasculature.
Results
Yes regulates shear stress-induction of pVE-cadherin
We investigated the pattern of VE-cadherin phosphorylation (pVE-cadherin) at Y685 in an intact vascular network (whole-mounted developing mouse retina at postnatal day 6; P6) in relation to the relative wall shear stress (WSS) level. The specificity of the pY685 antibody was validated in VE-cadherin tyrosine to phenylalanine, Y685F, mutant mice (Extended Data Fig. 1a). A similar strategy was used to validate the specificity of another pVE-cadherin antibody against pY731 (Extended Data Fig. 1b,c). Shear stress in different regions of the retina vasculature was estimated using computational flow simulation on the PolNet platform31 (Fig. 1a,b), which models flow based on the vascular plexus geometry and the rheological properties of blood. The retinal vasculature was segmented into five regions: sprouting front (region 1); vein (region 2); capillaries near vein (region 3); capillaries near artery (region 4); and artery (region 5) (Fig. 1b). Simulation showed that shear stress was the lowest at the sprouting front, correlating to a very low pVE-cadherin signal. In veins and capillaries (regions 2–4), with low-to-medium shear stress levels, the pVE-cadherin signal was markedly induced. In the artery, with very high shear stress, the pVE-cadherin signal returned to low levels (Fig. 1b), in accordance with findings in organs such as the diaphragm, the bladder and the yolk sac18,32.

a, Whole-mount staining of VE-cadherin (left) and pY685 VE-cadherin (middle) and computer-simulated WSS (right) modeling in the mouse P6 retina. An arterial branch (boxed) is shown in the enlarged pictures below. A, artery; V, vein. Scale bar, 200 µm. b, Segmentation of the retina into five regions (top) and plot (bottom) of regional WSS levels (right y axis; green bars) and corresponding pY685 VE-cadherin levels (left y axis; pink bars) in the different regions. c, Immunostaining of VE-cadherin (magenta) and pY685 VE-cadherin (green) in HUVECs cultured in static, 3 dyn cm−2 and 20 dyn cm−2 conditions for 24 h. Scale bars, 50 µm. d, Ratio of integrated intensity of pVE-cadherin/total VE-cadherin; n = 3 independent experiments. e, Representative immunoblot of pY685 VE-cadherin, total VE-cadherin, pY418 SFK, Src, Yes and GAPDH (for normalization) in static or shear stress-treated HUVECs; n = 3 independent experiments. f,g, Ratios of pY685 VE-cadherin/total VE-cadherin and pY418 SFK/GAPDH normalized to static conditions, n = 3 independent experiments. h, Immunofluorescent staining to show localization of Src and Yes (green) in HUVECs; co-stained for VE-cadherin (magenta). Scale bars, 50 µm. i, Relative expression levels of SRC and YES1 in HUVECs by qPCR. Comparison based on standard qPCR curves for Src and Yes transcripts from 10–2, 10–1, 1, 10, and 100 ng RNA. n = 3 independent experiments. j, Immunofluorescence showing pY685 VE-cadherin (green) and total VE-cadherin (magenta) in HUVECs transfected with control, YES1 or SRC short interfering RNA (siRNA) followed by exposure to 3 dyn cm−2 shear stress for 24 h. Scale bars, 50 µm. k, Quantification of pY685 VE-cadherin integrated intensity normalized to total VE-cadherin. n = 3 independent experiments. l, Representative immunoblots of pY418 SFK, Yes and Src in static or shear stress (3 dyn cm−2) treated control, YES1 or SRC-silenced HUVECs; n = 4 independent experiments. Quantifications of pY418 SFK normalized to GAPDH in different conditions are shown in m, n = 4 independent experiments. Bar graphs show mean ± s.e.m.; two-tailed Student’s t-test.
A similar pattern was seen in vitro; exposure of human umbilical vein endothelial cells (HUVECs) to low shear stress (3 dyn cm−2)-induced pY685 VE-cadherin levels, whereas at high shear stress (20 dyn cm−2), VE-cadherin was unphosphorylated (Fig. 1c,d). The appearance of pVE-cadherin in HUVECs correlated to phosphorylation of Src family kinases (SFKs) at Y418, indicative of SFK activity, which was induced under low shear stress and suppressed under high shear stress (Fig. 1e–g). A direct relationship between Src activation and phosphorylation of VE-cadherin has been suggested17; however, the amino acid sequence around Y418 in the kinase domain of Src is identical in multiple SFKs. Yes, another SFK expressed by ECs, was preferentially localized to EC junctions, whereas Src displayed a weak junctional signal and broad perinuclear and nuclear distribution (Fig. 1h). Moreover, YES1 transcript levels were higher than SRC levels in HUVECs (7.8 ± 0.5-fold; Fig. 1i). The expression of both Src and Yes was further upregulated by low shear stress (Fig. 1e).
To compare the roles of Yes and Src in the regulation of VE-cadherin phosphorylation, YES1- or SRC-silenced HUVECs were subjected to 3 dyn cm−2 shear stress. Silencing of either YES1 (siYES1) or SRC (siSRC) did not affect the expression of the other (Extended Data Fig. 1d,e); however, siYES1 strongly suppressed (82%) flow-induced phosphorylation of VE-cadherin at Y685 and Y658 (Fig. 1j,k and Extended Data Fig. 1f,g), whereas siSRC resulted in a partial loss (58%) of pY685 levels (Fig. 1j,k). Treatment of HUVECs exposed to 3 dyn cm−2 flow with siYES1 moreover suppressed flow induction of the pY418 SFK signal, whereas treatment with siSRC was without effect (Fig. 1l,m), indicating that Yes but not Src, phosphorylates VE-cadherin at low shear stress. Single-cell RNA-sequence analysis of P6 and P10 retinal ECs showed overall higher expression of Yes1 than Src in vivo; however, the expression of both was relatively uniform between non-proliferative vessel subtypes (Extended Data Fig. 1h,i and Supplementary Table 1), whereas expression of VE-cadherin trended toward higher levels in arteries (Extended Data Fig. 1j). Combined, these data suggest that the different levels of VE-cadherin phosphorylation in arteries and veins (Fig. 1a,b) are not caused by different expression levels of Yes1, but by different extents of flow-induced Yes kinase activity.
VE-cadherin phosphorylation on Y658, Y685 and Y731 by Yes
A conditional, Cdh5CreERT2-controlled33 Yes1 knockout mouse (Yes1 iECKO; Extended Data Fig. 2a) was generated to eliminate endothelial expression of Yes1. The deletion efficiency with tamoxifen treatment was 70% (Extended Data Fig. 2b). In the Yes1 iECKO P6 retina, phosphorylation of VE-cadherin was downregulated by 74 ± 15%, 78 ± 10% and 72 ± 23% at Y658, Y685 and Y731, respectively, compared to littermate controls (Fig. 2a–f and Extended Data Fig. 1a–c for pY685 and pY731 antibody specificity; the pY658 antibody has been described18). In EC-specific Src knockout (Src iECKO; 80% deletion efficiency28) pVE-cadherin levels decreased by 49 ± 15% (Y658), 39 ± 6% (Y685) and 46 ± 7% (Y731) in the P6 retina (Fig. 2g–i and Extended Data Fig. 2c–e). In Yes1 iECKO mice, pVE-cadherin levels were dramatically reduced in large veins, such as the vena cava (Extended Data Fig. 2f,g). Moreover, losing only one allele of Yes1 in ECs led to 60% decrease in pY658 and pY685 VE-cadherin levels in P6 Yeswt/fl:Cre+ retinas (Extended Data Fig. 2h,i). The long-term effect of endothelial Yes deficiency was examined in P22 mice, treated with tamoxifen at P1–3. The loss in VE-cadherin phosphorylation seen at P6 was still evident 3 weeks after recombination (Extended Data Fig. 2j,k), thus, other SFKs did not compensate for Yes1 deficiency during this time period.

a–c, Immunofluorescence of Yes1 iECKO (Yes1fl/fl, Cdh5CreERT2+) and control (Yes1fl/fl, Cdh5CreERT2−) P6 retinas showing total VE-cadherin (magenta) and phosphorylated VE-cadherin (green) for pY685 (a), pY658 (b) and pY731 (c). Scale bars, 50 µm. d–f, Quantification of pVE-cadherin (pY685) (d), pVE-cadherin (pY658) (e) and pVE-cadherin (pY731) (f) levels normalized to total VE-cadherin in control and Yes1 iECKO littermate P6 retinas. Control, n = 5; Yes1 iECKO, n = 4. g–i, Quantification of pVE-cadherin levels normalized to total VE-cadherin in P6 retinas of Src iECKO (Srcfl/fl, Cdh5CreERT2+) and control (Srcfl/fl, Cdh5CreERT2−) mice. pVE-cadherin (pY685) (g). Control, n = 8; Src iECKO, n = 10. pVE-cadherin (pY658) (h). Control, n = 5; Src iECKO, n = 5. pVE-cadherin (pY731) (i). Control, n = 7; Src iECKO, n = 10. j, Immunofluorescence showing the correlation between levels of pY685 VE-cadherin and YFP+ Yes1-deficient ECs in a P6 retina with chimeric recombination (100 µg tamoxifen per mouse) at P3. Arrows, YFP− ECs; arrowheads, YFP+ ECs. Scale bar, 20 µm. k, Quantification of pY685 VE-cadherin levels in YFP− and YFP+ ECs. n = 5 mice. l, Immunofluorescent images showing Yes (yellow), VE-cadherin (magenta) and pY685 VE-cadherin (green) in control (H11-STOP-Yes10/0, Cdh5CreERT2+; top) and Yes1 iECOE (bottom) retinas at P6. Arrows indicate arteries. Dashed lines indicate sprouting front. Scale bar, 200 µm. m, Quantification of pY685 VE-cadherin in the front and middle parts of control and Yes1 iECOE retinas. Control, n = 12; Yes1 iECOE, n = 8. n, Immunofluorescent staining of pY685 VE-cadherin in Yes1-overexpressing ECs in a P6 Yes1 iECOE retina with chimeric recombination. Arrows indicate ECs with Yes1 overexpression and arrowheads indicate ECs without recombination. Scale bar, 20 µm. o, Quantification of pY685 VE-cadherin in Yes1-overexpressing ECs and their neighboring non-recombined ECs, as shown in n, n = 7 mice. Bar graphs show mean ± s.e.m.; two-tailed Student’s t-test.
To study the cell-autonomous function of Yes1, the Rosa26R-YFP reporter was introduced into the Yes1 iECKO mouse line. One dose of tamoxifen at P3 generated chimeric recombination, labeling most, but not all, of the Yes1-deficient ECs with YFP. YFP+ retinal ECs showed 45 ± 12% lower pY685 VE-cadherin levels than adjacent YFP- ECs (Fig. 2j,k), indicating that Yes controls VE-cadherin phosphorylation in a cell-autonomous manner. The relatively limited reduction on pY685 VE-cadherin levels was expected as expression of the YFP reporter does not completely correlate with the Yes1 gene deletion.
VE-cadherin is a substrate for the EC-specific phosphotyrosine phosphatase VE-PTP (PTPRB)24,34. Silencing of PTPRB in HUVECs markedly increased pY685 VE-cadherin levels (Extended Data Fig. 3a–c). Treatment of HUVECs with siYES1 suppressed pY685 levels also in PTPRB-deficient ECs, indicating that Yes acts directly on VE-cadherin, rather than indirectly, by modulating VE-PTP activity.
Next, an inducible ‘floxed-STOP’ Yes1 overexpression mouse (denoted Yes1 iECOE) was generated by insertion of the mouse Yes1 complementary DNA with an upstream floxed transcriptional stop signal into the Hipp11 (H11) locus, controlled by the CAG promoter (Extended Data Fig. 4a). Following crossing with the Cdh5CreERT2 mouse line and treatment with tamoxifen, EC-specific Yes1 overexpression was achieved (H11-STOP-Yes1+/0, Cdh5CreERT2+). The overexpressed Yes protein localized to endothelial junctions in the retinas of Yes1 iECOE mice (Extended Data Fig. 4b), accompanied by a strong increase in the pY685 VE-cadherin signal. Strikingly, the pY685 VE-cadherin levels were increased at the sprouting front as well as in arteries in Yes1 iECOE P6 retinas to levels comparable to those in veins and capillaries (Fig. 2l,m). In mice with chimeric induction of Yes1 overexpression, pVE-cadherin levels increased several-fold also in retinal capillaries and veins where levels were already high in the control (Fig. 2n,o). This indicates that in wildtype (WT) mice, pVE-cadherin levels relative to total VE-cadherin protein is limited. Thus, assuming that VE-cadherin in the Yes1 iECOE mouse is fully phosphorylated, less than 35% of the total VE-cadherin pool was phosphorylated in veins and capillaries in the WT. We conclude that the limiting factor in flow-regulated VE-cadherin phosphorylation is the expression level of Yes, which when sufficiently high, allows its activation and phosphorylation of VE-cadherin also under conditions of high shear stress such as in arteries. Notably, in the chimeric Yes1 iECOE mouse retina, branch points on both veins and arteries were more likely to be occupied by Yes1 OE cells (Extended Data Fig. 4c,d), suggesting that ECs with high pVE-cadherin may be more resilient to disturbed flow at vessel bifurcations (note wall shear stress (WSS) changes at vessel branch points in Fig. 1a, right). Alternatively, Yes1 OE cells may be blunted in their ability to move away from bifurcations.
To compare the effects of Yes and Src overexpression on pY685 VE-cadherin, Src and Yes were introduced in HUVECs via lentiviral transduction. Forced overexpression of Src and Yes promoted increased pY685 VE-cadherin levels to a similar extent (Extended Data Fig. 5a–e); however, in Src-overexpressing cells, both Src and pY685 VE-cadherin were detected in the cytoplasm, concentrated in the perinuclear region, whereas in Yes overexpressing cells, both Yes and pY685 were detected at broad folds at endothelial junctions (Extended Data Fig. 5a; compare pY685 VE-cadherin immunostaining in Yes- and Src-overexpressing cells). These results suggest that Yes is the main kinase regulating VE-cadherin phosphorylation at EC junctions.
Yes1 controls endothelial collective cell migration
The plasticity of endothelial cell–cell adhesions is known to impact collective cell migration35. During vascular development, ECs migrate from veins toward arteries36. To explore whether Yes-mediated VE-cadherin phosphorylation impacts endothelial vein-to-artery migration, the dual reporter-Cre, iSuRe-Cre, was exploited for EC tracing37. In the iSuRe-Cre model, MdTomato-reporter expression is genetically linked with deletion of the target gene. The localization of MbTomato-labeled ECs in control and Yes1 iECKO retinas was determined at P7 and P15 after partial recombination induced at P3 (Fig. 3a). Artery and vein regions were segmented and the localization of MbTomato+ ECs was mapped in two-dimensions, shown with the relative distance between veins and arteries on the x axis (vein, 0.0; artery, 1.0) and the distance from optic nerve to sprouting front on the y axis (Fig. 3b). At P7, the vein-to-artery distribution of MbTomato+ ECs in Yes1 iECKO retinas was similar to the control (Fig. 3c–e); however, in the radial distribution, MbTomato+ ECs in Yes1 iECKO retinas were positioned closer to the optic nerve, compared to control MbTomato+ ECs (Fig. 3c,f,g). At P15, MbTomato+ ECs in Yes1 iECKO retinas were concentrated to arteries with only scarce presence in veins and capillaries (Fig. 3c–e). Moreover, at P15, Yes1 iECKO MbTomato+ ECs were localized closer to the optic nerve than in the control (Fig. 3c,f,g). With a higher degree of recombination following multiple tamoxifen injections at P1, 3, 6 and 9, the concentration of Yes-deficient ECs in arteries was striking (Fig. 3h,i), accompanied by widening of the artery diameter in the distal region, away from the optic nerve (Fig. 3i,j). Thus, compared to WT ECs, Yes-deficient ECs are privileged to migrate toward arteries in the developing retina. In retinas with chimeric deletion of VE-cadherin, certain arteries were entirely devoid of VE-cadherin-expressing ECs (Extended Data Fig. 6a), in agreement with the finding that loss of pVE-cadherin-dependent cell–cell adhesion promoted migration toward arteries38.

a, Timeline for EC distribution analysis (100 µg tamoxifen per mouse at P3). b, Schematics showing two-dimensional maps of fluorescently labeled ECs in flat-mounted retinas. x axis; relative distance between veins (0.0) and arteries (1.0); y axis; distance (µm) from optic nerve to sprouting front. c, Distribution analysis of ECs with Cdh5Cre-induced expression of MbTomato (green) in Yes1wt/wt, iSuRe-Cre+, Cdh5CreERT2+ and Yes1fl/fl, iSuRe-Cre+, Cdh5CreERT2+ at P7 (left) and P15 (right). CD31 (magenta) counterstaining shows all ECs. Boxed regions with veins and arteries shown enlarged (center). Distribution analysis (right) showing control (orange) and Yes1 iECKO (blue) ECs. Color density indicates average density of MbTomato+ cells in six retinas. Scale bars, 500 µm. d, Histogram plots of vein-to-artery distribution of MbTomato+ ECs in P7 and P15 retinas. Solid lines indicate median position of total MbTomato+ ECs detected. Dashed lines indicate positions of 25% and 75% of total MbTomato+ ECs. e, Box plot showing mean values of relative vein-to-artery distance of all MbTomato+ ECs/retina. Minima, maxima and center bounds show 25, 75 and 50 percentiles; whiskers show minimum and maximum values. n = 7 retinas for P15 Yes1 iECKO, n = 6 for all other groups; two-sided Welch’s t-test. f, Histogram plots showing radial distribution of MbTomato+ ECs in P7 and P15 retinas. Solid lines indicate median position of total MbTomato+ ECs detected. Dashed lines indicate positions of 25% and 75% of total MbTomato+ ECs. g, Box plot showing mean values of radial distance from optic nerve of all MbTomato+ ECs /retina. n = 7 retinas for P15 Yes1 iECKO, n = 6 for all other groups. Minima, maxima, center bounds and whiskers definitions and statistical analyses as in e. h, Timeline showing repeated tamoxifen administration. i, P15 retinas from Yes1wt/wt, iSuRe-Cre+, Cdh5CreERT2+ and Yes1fl/fl, iSuRe-Cre+, Cdh5CreERT2+ mice showing Cdh5Cre-induced expression of MbTomato+, normal and Yes1-deficient ECs (green); counterstaining for CD31 (magenta). Scale bars, 500 µm. j, Average artery diameter in proximal and distal positions (relative to optical nerve) in P15 retinas as shown in i. Control, n = 10 retinas; Yes1 iECKO, n = 5 retinas. Bar graphs show mean ± s.e.m.; two-tailed Student’s t-test. NS, not significant.
A monolayer scratch assay was employed to investigate how Yes deficiency affects EC migration in vitro. Individual cells from the first three rows from the migratory front were tracked and grouped by their initial position (Extended Data Fig. 6b and Supplementary Video 1). Control ECs in the different rows migrated collectively at a similar speed, whereas by silencing YES1, cells in rows 1–3 separated from neighboring cells and migrated at a faster speed (Extended Data Fig. 6c) and YES1-silenced cells from the first and second rows migrated further than the control cells (Extended Data Fig. 6d). Next, the activities of the well-known regulatory factors for migration, Rho GTPases CDC42, Rac1 and RhoA were examined (Extended Data Fig. 6e–h). The basal activity of CDC42 was increased in YES1-silenced cells. Basal RhoA activity showed the same trend, while Rac1 was unaffected, indicating that perturbed activity of small GTPases contributed to EC dysregulation in Yes1 deficiency39,40.
The loss of YES-dependent VE-cadherin phosphorylation and endothelial migration did, however, not interfere with sprouting angiogenesis. In Yes1 iECKO mice, retinal vascular outgrowth at P6 was normal compared to Cre+ control mice (Yes1wt/wt, Cdh5CreERT2+) (Extended Data Fig. 7a,b), and the number of filopodia on tip cells at the sprouting front was unaffected (Extended Data Fig. 7c,d). Retinal vascular outgrowth was unaffected also in mice carrying a Y685F VE-cadherin mutation (Extended Data Fig. 7e,f). In contrast, VE-cadherin-null mice (Cdh5 iECKO) exhibited a strong hypersprouting phenotype paralleled by delayed outgrowth (Extended Data Fig. 7g,h). Thus, the lack of VE-cadherin tyrosine phosphorylation in the Yes1 iECKO mouse does not result in a Cdh5 null phenotype. The retinal vascular area was reduced also in Yes1 iECOE mice compared to control (H11-STOP-Yes10/0, Cdh5CreERT2+) (Extended Data Fig. 7i,j). Therefore, VE-cadherin is strictly required, but its phosphorylation is dispensable for normal sprouting angiogenesis. Moreover, excessive phosphorylation of VE-cadherin is accompanied by disturbed vascular development.
We further investigated whether the lack of VE-cadherin phosphorylation would affect other cellular processes involved in angiogenesis. VEGFA induced angiogenic sprouting from aortic rings from both control and Yes1 iECKO mice (Extended Data Fig. 8a,b), but Yes1 iECKO sprouts were longer (Extended Data Fig. 8c), in accordance with enhanced cell motility. EC proliferation in developing retinas was assessed by 5-ethynyl-2-deoxyuridine (EdU) incorporation followed by immunostaining of the endothelial-specific nuclear protein ERG. The fraction of proliferative ECs was not changed in Yes1 iECKO mice compared to the control (Yes1fl/fl, Cdh5CreERT2−) (Extended Data Fig. 8d,e). Moreover, deleting Yes1 in ECs did not affect the number of Cleaved Caspase3-positive apoptotic cells in postnatal retinas (Extended Data Fig. 8f,g). These data show that Yes deficiency impacts collective EC migration while sprouting, proliferation and apoptosis still proceed.
Adherens junction morphology in Yes and Src deficiency
EC adaptation to microenvironmental demands for remodeling, perfusion or permeability is dependent on dynamic changes at adherens junctions, which appear with linear or jagged morphologies41 (Fig. 4a). In the WT P6 retina, VE-cadherin in arterial ECs formed linear junctions, whereas jagged junctions appeared only on veins and capillaries exhibiting phosphorylated VE-cadherin (Extended Data Fig. 9). YES1 silencing did not prevent formation of jagged junctions but the response to shear stress (3 dyn cm−2) was lower than in the control cells (Fig. 4b,c). In contrast, silencing of SRC in HUVECs exhibited a strong inhibitory effect on jagged junctions both in static and flow conditions (Fig. 4b,c). In P6 retinas, the appearance of jagged junctions in the veins of both Yes1 and Src iECKO mice was suppressed but more extensively so in the Src iECKO condition (Fig. 4d,e). These data show that changes in VE-cadherin morphology from linear to jagged does not directly correlate with its phosphorylation status.

a, Schematic outline of linear and jagged adherens junction morphology based on VE-cadherin immunostaining. b, Immunostaining of VE-cadherin in HUVECs transfected with control or YES1 or SRC siRNA and cultured under static or 3 dyn cm−2 flow conditions. Boxed regions show representatives of altered VE-cadherin morphologies, magnified to the lower right. Scale bars, 20 µm. c, Quantification of jagged junctions as shown in b, given as junction length in percentage of total. Bar graphs show mean ± s.e.m. with individual data points. siControl; n = 4, siYES1; n = 4, siSRC; n = 3 independent experiments. d, Adherens junction morphology in veins of retinal vessels at P6 illustrated by immunostaining of VE-cadherin (magenta) and pY685 VE-cadherin (green). Yellow arrowheads indicate jagged junctions. Scale bar, 10 µm. e, Quantification of jagged structures per 100 µm2 VE-cadherin area in control (Yes1fl/fl, Cdh5CreERT2−), Yes1 iECKO and Src iECKO mouse retinas. Control; n = 5 mice, Yes1 iECKO; n = 4 mice, Src iECKO; n = 6 mice. Bar graphs show mean ± s.e.m.; two-tailed Student’s t-test.
VE-cadherin internalization requires Yes, but not Src
In the siYES1-treated, flow-exposed HUVECs, VE-cadherin appeared in broad clusters along the junctions (Fig. 4b). The nature of these clusters was further explored by live cell imaging of HUVECs expressing green fluorescent protein (GFP)-tagged VE-cadherin. In control cells, VE-cadherin was detected in junction-proximal endocytic vesicles, which moved away from the junction. The internalized VE-cadherin vesicles disappeared within 2–5 min (Fig. 5a, top and Supplementary Video 2). In YES1-silenced HUVECs, VE-cadherin instead formed large clusters, which existed for 15–30 min before gradually disappearing, to be replaced by new clusters (Fig. 5a, middle and Supplementary Video 3). In SRC-silenced HUVECs, VE-cadherin endocytic vesicles were observed with similar dynamics as in control cells (Fig. 5a, bottom and Supplementary Video 4).

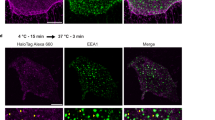
a, Static images from live imaging time series of HUVECs expressing green fluorescent protein (GFP)-tagged VE-cadherin. Boxed areas highlight examples of different dynamics of VE-cadherin and are shown enlarged (bottom right). Arrows for siControl (top) and siSRC (bottom) indicate internalized VE-cadherin vesicles. Arrows for siYES1 (middle) indicate a junctional VE-cadherin cluster, whereas arrowheads show detachment and internalization of VE-cadherin from the cluster. Time stamps are min:s. Scale bars, 10 µm. VE-Cad, VE-cadherin. b, Antibody feeding assay showing internalized VE-cadherin (magenta) in control, siYES1 and siSRC HUVECs co-stained with total VE-cadherin (green) and nuclei (blue). Scale bar, 20 µm. c, Quantification of internalized/total VE-cadherin in control, siYES1 and siSRC static HUVECs. Control; n = 8, siYES1; n = 4, siSRC; n = 4 independent experiments. d, Quantification of flow-induced internalized/total VE-cadherin in control, siYES1 and siSRC HUVECs. n = 3 independent experiments. Representative images are shown in Extended Data Fig. 10a. e, Quantification of internalized/total VE-cadherin in control, siYES1 and siSRC HUVECs treated with VEGFA or not for 3 h. n = 3 independent experiments. Representative images are shown in Extended Data Fig. 10b. f, Internalized VE-cadherin vesicles at the juxtamembrane regions in mouse vena cava ECs shown by immunostaining of VE-cadherin (magenta) and pY685 VE-cadherin (green). Cells highlighted in boxes are shown enlarged below. Arrowheads indicate internalized VE-cadherin vesicles. Scale bars, 20 µm. g, Quantification of internalized VE-cadherin vesicles/cells in control and Yes1 iECKO vena cava ECs. n = 3 mice for each group. Bar graphs show mean ± s.e.m.; two-tailed Student’s t-test.
These data indicated that VE-cadherin internalization was affected by Yes deficiency. VE-cadherin internalization was followed by an antibody feeding assay. Constitutive internalization of VE-cadherin was efficiently inhibited by YES1 silencing, whereas SRC silencing had no effect (Fig. 5b,c). HUVECs cultured under flow (3 dyn cm−2) showed increased VE-cadherin internalization, blocked by siYES1 but not siSRC treatment (Fig. 5d and Extended Data Fig. 10a). Additionally, VEGFA-induced VE-cadherin internalization was arrested by siYES1 but not siSRC (Fig. 5e and Extended Data Fig. 10b). VE-cadherin endocytic vesicles enriched in pY685 VE-cadherin were detected in vivo, just below the plasma membrane, in vena cava ECs from control mice. The prevalence of endocytotic vesicles was markedly reduced in the Yes1 iECKO vena cava (Fig. 5f,g). Combined, these results indicate that phosphorylation of VE-cadherin is strictly required for its constitutive internalization, which is a prerequisite for junctional plasticity.
p120-catenin has been implicated in the regulation of VE-cadherin endocytosis as binding of p120 masks an internalization motif in VE-cadherin14. The presence of the p120-catenin/VE-cadherin complex was assessed by coimmunoprecipitation, which showed similar levels under basal conditions and a trend toward increased complex formation in VEGFA-treated siControl and siYES1 HUVECs (Extended Data Fig. 10c,d). Thus, complex formation with p120 was unaffected by loss of VE-cadherin tyrosine phosphorylation.
Ultrastructure of Yes-deficient endothelial junctions
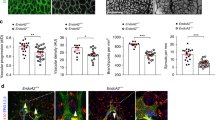
The decreased VE-cadherin internalization in Yes-deficient ECs could potentially impact junction organization, which was addressed by transmission electron microscopy (TEM) analysis of the mouse ear dermis subjected or not to intradermal injection of VEGFA. In Yes1 iECKO mice, the electron-dense junctional area expected to mark high actin density, known to be disrupted in conjunction with vascular leakage1,42,43, was smaller than that in control mice under basal conditions (Fig. 6a,c). While the electron-dense area decreased upon VEGFA injection in WT mice, it increased in the Yes1 iECKO condition (Fig. 6a,c), suggesting disturbed junctional dynamics under Yes1 deficiency. The electron-dense cortical actin area in Src iECKO mice was similar to control mice with a trend toward decreased area with VEGFA treatment (Fig. 6b,d).

a,b, Endothelial junctions in control, Yes1 iECKO or Src iECKO mouse ear dermis vessels after intradermal injection of saline or VEGFA164, visualized by TEM. EC junction length is highlighted in green and electron-dense junctional area (cortical actin-rich domain) is in red. Scale bars, 500 nm. c, Quantification of actin area/junction length in control (Yes1fl/fl, Cdh5CreERT2−) and Yes1 iECKO mice. Saline-treated control mice, n = 9; VEGFA-treated control mice, n = 7; saline-treated Yes1 iECKO mice, n = 10; VEGFA-treated Yes1 iECKO mice, n = 11. Overall, 2–40 junctions were analyzed in each mouse. d, Quantification of electron-dense junctional area/junction length in control (Srcfl/fl, Cdh5CreERT2−) and Src iECKO mice. n = 5 mice for each group. e, Staining with phalloidin shows the arrangement of actin stress fibers and cortical actin in HUVECs treated with control, YES1 or SRC siRNA followed by VEGFA stimulation for 15 min. Scale bars, 20 µm. f, Quantification of the ratio of cortical actin/stress fiber in control, YES1- or SRC-silenced HUVECs. n = 3 independent experiments. Bar graphs show mean ± s.e.m.; two-tailed Student’s t-test.
In agreement, live imaging of cells labeled with fluorescent actin revealed that actin stress fibers were misaligned and moved toward the periphery in YES1- but not in Control- or SRC-silenced HUVECs (Supplementary Videos 5–8). Moreover, VEGFA treatment increased the relative abundance of cortical actin in YES1-silenced HUVECs but not in SRC-silenced or control cultures. In the siSRC cultures, there was instead an increased proportion of stress fibers relative to cortical actin (Fig. 6e,f) in accordance with the different ultrastructural TEM morphologies of Yes1 iECKO and Src iECKO vessels.
Increased vascular permeability in Yes1 iECKO mice
The consequence of Yes deficiency for vascular barrier function was addressed using several in vivo methods. The basal level of vascular permeability was tested by tail vein injection of Evans blue followed by a 2-h circulation period. Increased levels of Evans blue in the interstitial tissue of the ear skin were detected in Yes1 iECKO mice (Extended Data Fig. 11a). Blood–brain barrier integrity, assessed by the leakage of Evans blue and cadaverine into the cerebral tissue, was unaffected by Yes1 deficiency (Extended Data Fig. 11b,c). Thus, the vascular barrier was compromised in the Yes-deficient dermis and potentially other tissues but not in the CNS.
The permeability of developing retinal vessels was studied by cardiac injection of fixable fluorescent tracer (dextran-TRITC, 10 kDa) in P6 mice. Leakage of the tracer into the perivascular tissue was observed preferentially at the vascular sprouting front, possibly marking an immature vascular barrier, and neither Yes1 deletion nor overexpression changed the number of leakage sites at the sprouting front (Fig. 7a,b); however, an increased frequency of venous leakage was observed in Yes1 iECKO retinas (eight leakage sites observed in 11 veins from ten retinas) in comparison to the control retinas (two leakage sites in 14 veins from nine retinas) (Fig. 7a,c). In contrast, leakage from veins was not elevated in Yes1 iECOE retinas (Fig. 7c).

a, Perivascular leakage of 10 kD dextran (magenta) in fixed P6 retinas of control (Yes1fl/fl, Cdh5CreERT2−), Yes1 iECKO and Yes1 iECOE mice. Vessels shown by CD31 immunostaining (green). Boxed leakage sites shown in magnification. Scale bars, 200 µm. b, Quantification of leakage sites at the sprouting front. Control, n = 9 retinas; Yes1 iECKO, n = 10 retinas; Yes1 iECOE, n = 6 retinas, two-tailed Student’s t-test. c, Quantification of leakage sites in retinal veins. Control, n = 14 veins per 9 retinas; Yes1 iECKO, n = 11 veins per 10 retinas; Yes1 iECOE, n = 7 veins per 6 retinas; two-tailed Student’s t-test. d, Static images from intravital time series imaging of mouse ear dermal vessels before and 10 min after intradermal VEGFA injection. Leakage visualized by the perivascular accumulation of 2,000 kDa dextran (magenta). Scale bars, 50 µm. e, Heat map images of individual leakage sites (arrows) after VEGFA injection. Scale bars, 20 µm. f, Quantification of individual leakage sites at veins and capillaries in control and Yes1 iECKO mice induced by VEGFA. Control, n = 5 mice and Yes1 iECKO, n = 4 mice. g, VEGFA-induced changes in perivascular fluorescence intensity measured at 2-s intervals for 1,500 s. Curves show mean values of eight leakage sites from five control mice and ten sites from four Yes1 iECKO mice; two-way analysis of variance (ANOVA). h, Quantification of individual leakage sites at veins and capillaries of control (H11-STOP-Yes10/0, Cdh5CreERT2+) and Yes1 iECOE mice induced by VEGFA. Control, n = 7 mice, Yes1 iECOE, n = 6 mice. i, Quantification of individual leakage sites at veins and capillaries in the ear dermis of control (Srcfl/fl, Cdh5CreERT2−) and Src iECKO mice after VEGFA injection. Control, n = 6 mice, Src iECKO, n = 6 mice. j, VEGFA-induced changes in perivascular fluorescence intensity measured at 2-s intervals for 1,500 s. Control, n = 6 mice, Src iECOE, n = 6 mice; two-way ANOVA. k, FACS analysis of monocyte/macrophage (CD11b+, LY6G−) extravasation in peritoneal fluid from control and Yes1 iECKO mice 24 h after intraperitoneal (i.p.) injection with saline or VEGFA, n = 7 mice for each group. Bar graphs show mean ± s.e.m.; two-tailed Student’s t-test.
Permeability of the mature vasculature was investigated by intravital confocal imaging of adult dermal vessels. In the WT dermis, pVE-cadherin was detected in some but not all venous vessels and, unexpectedly, in some arterioles (Extended Data Fig. 11d). This is in contrast to the pattern observed in the retina, where veins but not arteries display pVE-cadherin signals. The detection of pVE-cadherin in dermal arterioles may reflect flow patterns that do not conform to the expected high-flow–low-phosphorylation pattern or alternatively, higher expression levels of VE-cadherin. Nevertheless, VE-cadherin phosphorylation in dermal vessels was markedly reduced in adult Yes1 iECKO mice (Extended Data Fig. 11d,e), similar to the findings in the developing retina. To monitor leakage, a fluorescent tracer was injected into the tail vein and leakage from dermal vessels in the ear was monitored after local injection of VEGFA (Fig. 7d and Supplementary Videos 9 and 10). Leakage was initiated at individual sites in venules and capillaries in both control and Yes1 iECKO mice (Fig. 7e and Supplementary Videos 11 and 12). Quantification of leakage sites/vessel length showed that the leakage site density was increased in both venules and capillaries in Yes1 iECKO mice (Fig. 7f). The fluorescence intensity profile at leakage sites demonstrated that leakage appeared sooner after VEGFA injection in Yes1 iECKO mice and that leakage was more vigorous in the absence of Yes1 compared to the WT (Fig. 7g). The number of leakage sites was unaffected in Yes1 iECOE mice compared to WT mice (Fig. 7h), indicating that leakage is not directly regulated by VE-cadherin tyrosine phosphorylation. In contrast to the increased leakage observed in Yes1 deficiency, Src iECKO mice exhibited a reduced number of leakage sites after VEGFA injection (Fig. 7i). The kinetics of the remaining leakage sites was unaffected (Fig. 7j). These results show that while Yes is required for restricting formation of leakage sites at junctions, Src is required for such sites to form.
The Y731 phosphosite in VE-cadherin is required for leukocyte extravasation. Leukocyte populations were analyzed by FACS in fluids collected by peritoneal lavage in mice 24 h after saline or VEGFA was injected. In agreement with the phenotype of Y731F mice11, monocyte (CD11b+LY6G−) extravasation was reduced in VEGFA-treated Yes1 iECKO mice compared to the control (Fig. 7k).

Yes protein-tyrosine kinase is inactive under the high shear stress in arteries. Low shear stress level in capillaries and veins activate Yes kinase activity, which phosphorylates the adherens junction protein VE-cadherin at Y658, Y685 and Y731. Phosphorylation of VE-cadherin is required for its constitutive internalization, which confers EC junctional plasticity. Junctional plasticity is essential for collective EC migration during angiogenesis as well as for maintaining vascular barrier integrity.
Combined, these results show that loss of Yes-mediated phosphorylation of VE-cadherin results in dysregulation of endothelial junctions with increased leakage of macromolecules but suppressed extravasation of inflammatory cells.
Discussion
Regulation of endothelial junctions has been ascribed to Src44,45. The viral form, v-Src, localizes to adherens junctions46 and gap-junction-dependent cell–cell communication is regulated by c-Src47. Although broad screens for Src kinase substrates have revealed hundreds of candidate molecules48, only a handful have been validated, including p85-Cortactin, p110-AFAP1, p130Cas, p125FAK and p120-catenin27. A common denominator for several of these Src substrates is the involvement in actin-dependent processes. In agreement, EC-specific loss of Src expression results in impaired cell-matrix adhesion properties28.
Even less is known about Yes substrates. Yes phosphorylates the tight junction component occludin in epithelial cells and ECs49, but the functional consequence is unclear. Yes has been implicated in negative regulation of the cell cycle by phosphorylating cyclin-dependent kinase 4 (ref. 50). Yes moreover plays a role in T-lymphocyte immunity by phosphorylating CD46 (ref. 51) and collapsin response mediator protein 2 (CRMP2)52; however, using an in vitro kinase-based screen to identify substrates for SFKs, no specific substrate for Yes was identified53. The important role of Yes in EC cell–cell adhesion is suggested by its abundant junctional localization; single-cell transcriptomics shows that Yes1 is more abundant in ECs than in other cell types in a range of mouse tissues (https://tabula-muris.ds.czbiohub.org/)54.
A notable conclusion is that the different SFKs must be targeted individually as they serve different, sometimes opposing purposes. Yes deficiency resulted in loss of VE-cadherin phosphorylation, suppressed endocytosis, clustering of VE-cadherin and increased macromolecular leakage. In contrast, in Src deficiency, VE-cadherin phosphorylation was reduced, albeit to a lesser extent, VE-cadherin internalization remained unaffected and macromolecular leakage was suppressed. Combined, these data challenge the cause–consequence relationship between VE-cadherin phosphorylation, internalization and the formation of junctional gaps and macromolecular leakage. Specifically, the loss of the Y685 site has been shown to prevent tracer leakage in healthy and pathological tissues11,22,55. Furthermore, VEGFA- or histamine-induced acute vascular leakage in mature dermal vessels is established at pVE-cadherin-high sites in capillaries and postcapillary venules5,56. Combined, the data suggest that VE-cadherin phosphorylation is required for its constitutive endocytosis and it sensitizes ECs to rapid VEGFA-induced endocytosis and gap formation at EC junctions (see schematic outline, Fig. 8). Nevertheless, an additional trigger is required for gap formation and macromolecular leakage. Of note, phosphorylation of VE-cadherin could also be a prerequisite for gap closure. Thus, the dysregulated leakage from Yes-deficient vessels may be a consequence of uncoordinated opening and closure of junctional gaps in addition to incomplete sealing in the resting state due to loss of plasticity, akin to a stiff rubber band that has lost its normal elasticity.
The mechanism underlying the distinct effects of Yes and Src on VE-cadherin may be related to their different subcellular localization, allowing phosphorylation of different subcellular pools of VE-cadherin, in addition to the potential involvement of other kinase-specific substrates. Src localizes to focal adhesions in freshly seeded ECs, where it phosphorylates p125FAK27. Endothelial FAK-deficiency results in dismantling of adherens junctions57, and VEGFA-induced vascular leakage requires FAK-induced phosphorylation of β-catenin58, underscoring the important role of matrix adhesion in the regulation of EC junctions. The subcellular localization of cytoplasmic tyrosine kinases is influenced by NH2-terminal fatty acid modification. Both Yes and Src are myristoylated, whereas Yes is also palmitoylated, potentially contributing to steer the subcellular localization and trafficking of Src and Yes59,60. Their different subcellular localization would also be compatible with the distinct regulation of the actin cytoskeleton by Yes and Src; Yes- but not Src-deficient HUVECs exhibited an increase in cortical actin and loss of actin stress fibers. Moreover, the electron-dense, cortical actin-enriched junctional area in Yes-deficient dermal vessels was low in the basal condition but increased markedly upon administration of VEGFA. The abnormal actin arrangements in the absence of Yes provide a potential mechanistic underpinning for the changes in junctional dynamics with formation of VE-cadherin clusters and the block in VE-cadherin internalization.
Binding of p120-catenin to a VE-cadherin juxtamembrane motif has been implicated in VE-cadherin stabilization14, and in agreement, dephosphorylation of VE-cadherin at Y658 increases the binding affinity for p120 in vitro19,61. The vascular barrier is normal in mice with a mutation of this endocytic motif in VE-cadherin, but the vessel density is decreased in the developing retina62. VE-cadherin/p120 complex formation occurs, however, independently of VE-cadherin phosphorylation18. In accordance, p120/VE-cadherin complex formation occurred also in the absence of Yes. Notably, in contrast to certain VE-cadherin endocytosis-deficient mutant models62, the expression levels of VE-cadherin and p120 were unaffected by Yes1 deletion. Moreover, YAP/TAZ signaling is implicated in EC adhesion and VE-cadherin internalization63; the potential contribution of Yes to YAP/TAZ signaling warrants further studies.
The exaggerated macromolecular leakage in Yes1−/− vessels is at odds with the leakage phenotype of Y685F mice, which show suppressed leakage of macromolecules11,22 and suppressed reverse transmigration of neutrophils21. The difference in phenotypes between the Y685F- and Yes-deficient mouse models may be related to the fact that Yes deficiency affects additional molecular regulators at endothelial junctions yet to be discovered; however, the block in monocyte extravasation is in agreement with the phenotype of the Y731F vasculature11. Thus, although the phosphosites are similarly regulated by Yes, Y658, Y685 and Y731 serve distinct purposes by regulating the extravasation of either macromolecules or inflammatory cells through different mechanisms and potentially at geographically distinct sites in the vasculature.
The new insights presented here deepen the understanding of the complex regulation of adherens junctions, where an interplay between several cytoplasmic tyrosine kinases, such as Yes and Src, orchestrates the process when gaps form and close again and when the vascular surface becomes prepared to dock and expel inflammatory cells.
Methods
Mice
The Yes1flox/flox mouse strain was generated by flanking exon 3 with loxP sites, introduced by homologous recombination on the genetic C57BL/6 background (Taconic/Cyagen; Extended Data Fig. 2a). The generation of Srcflox/flox and Cdh5flox/flox strains has been described28,55. These strains were crossed with Cdh5(PAC)-CreERT2 mice (a kind gift from R. Adams) to generate endothelial-specific knockout of Yes1, Src or Cdh5. For analysis in mosaic knockout, the fluorescent reporter mouse line B6.129×1-Gt(ROSA)26Sortm1(EYFP)Cos/J (stock no.006148, The Jackson Laboratory) was introduced to indicate Cre activity with YFP expression. For retinal EC distribution analysis, the iSuRe-Cre mouse strain (a kind gift from R. Benedito) was crossed with the Yes1flox/flox mouse. iSuRe-Cre+; Yes1wt/wt; Cdh5CreERT2+ mice were used as control for all analyses of the iSuRe-Cre+; Yes1fl/fl; Cdh5CreERT2+ strain, to ensure that the observed phenotype was not due to Cre toxicity. Conditional Yes1 knock-in mouse H11-CAG-STOP-Yes1 was generated using CRISPR/CAS (Taconic/Cyagen; Extended Data Fig. 4a). The VE-Cad Y685F and the VE-Cad Y731F mice have been described11. Both males and females were included in the experiments. Animal husbandry and procedures were in accordance with institutional guidelines and approved by the Institutional Review Board for animal experimentation at Uppsala University (permit no. 5.8.18–06789/2018).
Intravital vascular leakage assay
Intravital imaging of the mouse ear dermis5 was performed following systemic administration of 2,000 kDa dextran by tail vein injection. Mice were anesthetized by i.p. injection of ketamine-xylazine (120 mg kg−1 ketamine, 10 mg kg−1 xylazine) to a surgical level, and the ear was secured to a solid support. Mice were maintained at a body temperature of 37 °C throughout the experiment for a maximum 90 min. Timelapse imaging was performed using single-photon microscopy (Zeiss LSM 710) and a high NA water-immersion lens (CF175 apochromat ×25 W NA 1.1, Nikon). For intradermal EC stimulation, a volume of 0.1 μl recombinant mouse VEGFA164 (Peprotech), 100 ng μl−1 was injected using a submicrometer capillary needle. Then, 10 kDa TRITC dextran was used as a tracer for VEGFA. Leakage sites were identified in timelapse imaging following VEGFA injection as defined sites of concentrated dextran in the extravascular space. The researcher was blinded to the genotype of the mice when performing the analyses.
In vivo wall shear stress analysis
Flow simulation from the images of retinal vasculature shown by CD31 staining was performed using the PolNet platform31. Flat-mounted retinas were imaged at an area including one artery, one or two veins and the complete capillary network in between by tile scanning confocal microscopy (Leica SP8) using a ×40 objective. Three-dimensional reconstruction of the vascular network was performed using maximum intensity projection of the vessels illustrated by CD31 immunostaining. A heat map of the relative shear stress level was generated based on the flow simulation by assigning an inlet at the artery and outlet at the vein. Regional average shear stress levels were obtained and plotted combined with the pVE-cadherin levels.
Retinal EC distribution analysis
Chimeric recombination was induced in iSuRe-Cre+ mice at P3 by i.p. injection of tamoxifen (100 µg per mouse, Sigma). Retinas were taken at P7 and P15, immunostained for CD31 and flat-mounted. Images were taken by z-stack tile scanning using a ×10 objective on a confocal microscope (Leica SP8). Maximum intensity projection images of whole retinas were used for image segmentation, which was performed with ImageJ resources. The maximum projection of the MbTomato channel threshold was established to distinguish MbTomato+ cells from the background. Outliers with a radius between 0.2–1.0 µm were removed. The CD31 channel (after maximum projection) was used to define the outlines of veins and arteries; the optic nerve was used as a mask to define a referential system. For computational analysis, a bespoken Python-based workflow was employed, accessible on GitHub (https://github.com/wgiese/retina-vein-artery-cs). For every pixel in the image, three numbers were computed (using the mask as referential): (1) distance to the nearest vein (dv); (2) distance to the nearest artery (da); and (3) radial distance to the optic nerve (r). From these measures, the relative distances were obtained by ϕv-a = dv/(dv + da). The EC distribution was computed by performing the operation for all MbTomato-positive pixels, which were used as a proxy for EC distribution. A kernel density estimation was used to approximate the underlying EC distribution in the two-dimensional coordinate system spanned by ϕv-a and r. High-degree recombination was induced at P1, P3, P6 and P9 by i.p. injection of tamoxifen (100 µg per mouse per day, Sigma) and retinas were taken at P15 for analysis.
Retinal single-cell RNA-sequencing data
Single-cell RNA-sequencing data generated from WT P6 and P10 retinas64 was obtained from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) accession no. SRP322112 and preprocessed with CellRanger (v.5.0.1) using the mm10 reference genome. ECs included in the analysis were defined as positive for Pecam1, Cdh5 or Kdr expression or as having been characterized as ECs in the original publication. Raw counts were normalized with a pooling size factor-based strategy as implemented in scran (v.1.18.7). Subtype annotations were generated using scmap (v.1.16.0) using the cluster assignment described in the original publication. Differential gene expression analysis was carried out using MAST as implemented in Seurat (v.4.1.1). The effects of dropouts in the data were reduced with imputation with the magic Python package (v.0.1.1; k = 9, ka = 3, t = 1) and graphs were generated with ggplot2 (v.3.3.6).
Vessel leakage in postnatal retina
Recombination was induced at P1-3 by i.p. injection of tamoxifen (100 µg d−1 per mouse, Sigma). At P6, mice were anesthetized, and 10 kDa TRITC dextran-lysine (100 µg g−1 body weight, TdB Labs) was injected into the left heart ventricle. After 10 min, eyes were removed and fixed in 4% paraformaldehyde (PFA; Sigma) for 2 h at room temperature (RT). A needle puncture on the cornea facilitated fast fixation. Retinas were immunostained with CD31 to visualize blood vessels. Images were acquired by z-stack tile scanning in a confocal microscope (Leica SP8). A site of leakage was scored positive if dextran was detected in the perivascular region.
Immune cell infiltration assay
Recombination was induced in 8–12-week-old mice by gavage with tamoxifen (2 mg d−1 per mouse, Sigma) for 5 consecutive days. After 2 d resting, saline or VEGFA (5 µg kg−1 body weight) was administered through i.p. injection. After 24 h, mice were killed, 5 ml of PBS was i.p. injected and recruited cells were collected after abdominal palpation. Cells were immunostained with CD11b (101206, BioLegend) and LY6G (127608, BioLegend) antibodies for FACS analysis.
Transmission electron microscopy
Eight to 12-week-old control or Yes1 iECKO mice were treated with intradermal injection of VEGFA164 (10 ng, PeproTech) in the ear skin. The other ear served as an untreated control. Mice were anesthetized and perfused first with 10 ml Hank’s balanced salt solution and then 12 ml cold fixative (1% glutaraldehyde (Sigma), 4% PFA in 0.1 M phosphate buffer) through the left ventricle. Ears were cut and placed in fixative for 30 min at 4 °C, washed three times with PBS, dehydrated and embedded in Epon, LX112 (Ladd Research Industries). Imaging was performed with a Tecnai G2 Spirit 120 kV transmission electron microscope (Fei Europe) and Quamesa CCD camera. Image analysis was done with Fiji processing package of Image J2 software.
Aortic ring assay
Thoracic aortas were taken from P7 mice and cut into 1 mm rings after removal of the connective tissues. Rings were embedded between two layers of rat tail collagen, type I (Thermo Fisher Scientific), and cultivated in Dulbecco’s modified Eagle’s medium (Gibco) supplemented with 10% fetal bovine serum (FBS) and VEGFA (PeproTech, 30 ng ml−1). Then, 4-OH-tamoxifen (5 µg ml−1, Sigma) was added to the culture for 48 h to induce recombination. After 4 d of initial cultivation, samples were fixed in 4% PFA (Sigma) and analyzed by microscopy after immunostaining.
Proliferation
EC proliferation was assessed by i.p. injection of EdU (100 µg per mouse, Thermo Fisher Scientific) at P6. Mice were killed 2 h after injection, and retinas were removed and fixed in 4% PFA (Sigma). EdU staining of retinas was performed using the Click-iT EdU imaging kit (Thermo Fisher Scientific). The samples were co-stained with antibodies against ERG to identify EC nuclei. Images were taken by confocal microscopy (Leica SP8) and analyzed by ImageJ.
Isolation of mouse lung ECs
Mice were treated with tamoxifen (100 µg d−1 per mouse, Sigma) at P1-3. Lungs were taken at P6 and dissociated to obtain cell suspensions using the MACS dissociation kit (Miltenyi Biotec). Isolation of ECs from mouse lungs was performed using anti-CD31 antibody (BD Pharmingen)-conjugated Dynabeads (Thermo Fisher Scientific). RNA from the isolated ECs was extracted immediately by using an RNeasy mini plus kit (QIAGEN).
Cell culture
HUVECs (PromoCell) were cultured at 37 °C, 5% CO2 in MV2 medium with supplied supplements (PromoCell). Cells were tested for cell morphology and cell-type-specific markers using flow cytometric analyses by the vendor. For VEGFA stimulation, confluent cells were starved in MV2, 0.2% FBS without supplements for 5 h. The same medium with VEGFA165 (50 ng ml−1, PeproTech) was added and cells were incubated at 37 °C for the indicated time periods.
Yes and Src overexpression in vitro
SRC cDNA was cloned into pLV-CMV-IRIS-PURO-mScarlet plasmid at restriction enzyme site XhoI using In-Fusion Cloning (Takara). YES1 cDNA was cloned into the pLV-CMV-IRIS-PUR-mScarlet plasmid at XhoI site using In-Fusion Cloning (Takara). Lentivirus constructs of control-m-Scarlet, c-Src-mScarlet, and Yes-mScarlet were packaged into lentivirus in HEK-293T cells by co-transfection of third-generation lentiviral packaging plasmids with PEI 2500 (BioScientific). Lentivirus containing supernatant was collected on day 2 and 3 after transfection. Lentivirus was concentrated by Lenti‐X concentrator (Clontech, 631232). Transduced target HUVECs were selected with puromycin (1 mg ml−1) after 24 h and used for assays after 72 h.
Scratch wound healing assay
HUVECs were seeded at a density of 50,000 cells per well into a 96-well ImageLockTM tissue culture plate (Essen BioScience) and incubated in MV2 medium overnight followed by standardized scratching using WoundMaker (Essen BioScience). Cells were washed with PBS, and the plate was placed into an IncuCyte ZOOM (Essen BioScience) and scanned every 15 min for 12 h using a ×10 objective, during which, data were collected using ZOOM software (Essen BioScience). Cellular migration was analyzed using MTrackJ in ImageJ by manually marking the cells at each time frame. The migration speed of one cell was obtained by dividing the migration distance by time.
Confocal live cell imaging
HUVECs were seeded at full confluency on glass-bottom plates (MatTek) and cultured in MV2 medium overnight. Fresh medium with SiR-actin dye (Cytoskeleton) was added to the cells and incubated for 2 h. The cell monolayer was scratched using a 2-µl pipette tip and changed to fresh medium. The plates were then placed on a Leica SP8 microscope equipped with a humidified CO2 incubator (Leica Microsystem). Timelapse z-stack images were scanned every 1 min for 3 h.
In vitro flow treatment
HUVECs were seeded at full confluency on µ-slides (Ibidi) coated with fibronectin (1 µg ml−1, Sigma) and cultured overnight in degassed MV2 medium. The slides were then connected to the Ibidi pump system to allow degassed medium to flow through at 3 or 20 dyn cm−2 shear stress for 24 h at 37 °C in a CO2 incubator. Static control cells were cultured on slides in an incubator for the same amount of time. Cells were fixed in 1% PFA for 10 min for immunofluorescence or lysed in RIPA buffer (Thermo Fisher Scientific) for western blots.
VE-cadherin internalization assay
Control or YES1-silenced confluent HUVECs were incubated with an antibody against the VE-cadherin extracellular domain (Clone BV6, MABT134, Merck Millipore, 10 µg ml−1) at 4 °C for 1 h in MV2 basal medium containing 3% bovine serum albumin (BSA) (Sigma) without supplements. Unbound antibody was removed by rinsing cells in ice-cold MV2 basal medium. Cells were then cultured at 37 °C for 4 h in the presence of 150 µM chloroquine (Sigma) in MV2 medium with supplements. For VE-cadherin internalization under flow, cells were first cultured under 3 dyn cm−2 flow for 20 h before adding the antibody (Clone BV6, MABT134, Merck Millipore, 10 µg ml−1) and chloroquine (150 µM) in the medium followed by continued culturing under flow for 4 h. Cells were then washed for 8 min with PBS containing 25 mM glycine and 3% BSA with Ca2+ and Mg2+ at pH 2.7, to remove antibody from the cell surface, followed by rinsing with PBS and fixation with 2% PFA for 10 min at RT. Total VE-cadherin was stained with an antibody generated in another species (AF1002, R&D Systems). Samples were mounted on slides with Fluoromount G (Southern Biotech) and analyzed by confocal imaging.
RNA interference
siRNAs targeting human YES1, SRC or PTPRB (Sigma) were transfected into HUVECs by using Lipofectamine RNAiMAX (Thermo Fisher Scientific). Control cells were transfected with control siRNA that did not target any specific gene. siRNAs were mixed with lipofectamine and added to the cells at a concentration of 10 nM and incubated overnight before changing to fresh medium. The cells were used for experiments 48 h after transfection. The knockdown efficiency was validated by quantitative PCR. YES1 siRNA: SASI_Hs01_00086922; SRC siRNA: SASI_Hs01_00112907; PTPRB siRNA: EHU158501, Sigma; Control siRNA: MISSION siRNA universal negative control (SCI001).
Immunofluorescence
Mice were killed and tissues were removed and fixed in 2% PFA (eyes and ears were fixed for 1 h, vena cava was fixed for 15 min). Samples were incubated in PBS, 0.5% Triton X-100, 1% BSA for 3 h at RT and then with primary antibodies overnight at 4 °C. After three washes at RT, samples were incubated with secondary antibodies overnight at 4 °C, washed three times and mounted on slides for imaging. For staining of cultures, cells were fixed in 1% PFA for 10 min after treatment. After fixation, the cells were washed three times in PBS and permeabilized in PBS, 0.1% Triton X-100 for 5 min, followed by blocking in 1% BSA for 30 min and incubation with primary antibodies overnight at 4 °C. Cells were washed three times in 0.1% Triton X-100, incubated with secondary antibodies for 1 h at RT, washed three times and mounted. Imaging was done using Leica SP8 (Leica Microsystems) confocal microscopes with LAS X software (v.3.5.7.23225). Image processing and analysis were performed using ImageJ software.
Antibodies against pVE-cadherin Y685 or Y731 were generated by immunizing rabbits with phosphopeptides of the corresponding regions of mouse VE-cadherin (New England Peptide). The pY658 antibody was a kind gift from E. Dejana, IFOM, Milano, Italy and Uppsala University). Antibodies were purified and precleared by incubation on fixed and permeabilized Cdh5 null mouse ECs before use. The commercial antibodies used were goat anti-mouse VE-cadherin (AF1002, R&D Systems, 1:500 dilution), mouse anti-VE-cadherin-alexa-647 (561567, Becton Dickinson, 1:500 dilution) goat anti-mouse CD31 (AF3628, R&D Systems, 1:500 dilution), chicken anti-GFP (ab13970, Abcam, 1:1,000 dilution), rabbit anti-ERG (ab92513, Abcam, 1:500 dilution), mouse anti-Yes (610376, BD Biosciences, 1:400 dilution), mouse anti-Src (Clone GD11, Merck Millipore, 1:400 dilution) and Alexa Fluor 647-phalloidin (A-22287, Thermo Fisher Scientific, 1:100 dilution). Secondary antibodies conjugated with Alexa Fluor dyes were obtained from Thermo Fisher Scientific or Jackson ImmunoResearch Laboratories.
Quantitative PCR
RNA was extracted and purified using RNeasy Plus kit (QIAGEN). RNA concentrations were measured by a Nanodrop spectrophotometer (Thermo Fisher Scientific) and adjusted to equal concentrations, followed by reverse transcription using SuperScript III (Thermo Fisher Scientific). Real-time quantitative PCR was performed on a Bio-Rad real-time PCR machine using SsoAdvanced qPCR reagent (Bio-Rad). The housekeeping gene ribosomal protein L19 (Rpl19) was used as an internal control. The comparative Ct method was used to calculate fold differences.
Primer sequences were as follows: mouse Yes1: forward: 5′-AGTCCAGCCATAAAATACACACC-3′, reverse: 5′-TGATGCTCCCTTTGTGGAAGA-3′; mouse Rpl19: forward: 5′-GGTGACCTGGATGAGAAGGA-3′, reverse: 5′-TTCAGCTTGTGGATGTGCTC-3′; human YES1: forward: 5′-CTCAGGGGTAACGCCTTTTGG-3′, reverse: 5′-CACCACCTGTTAAACCAGCAG-3′; human SRC: forward: 5′-GACAGGCTACATCCCCAGC-3′, reverse: 5′-CGTCTGGTGATCTTGCCAAAA-3′; human RPL19: forward: 5′-TCGCCTCTAGTGTCCTCCG-3′, reverse: 5′- GCGGGCCAAGGTGTTTTTC-3′; human PTPRB: forward: 5′-GCGGACCAGGATTCCCTCTA-3′, reverse: 5′-AACTCCCGGATGGTCC-3′.
Rho-GTPase activity assay
Control or YES1-silenced HUVECs were starved at 37 °C in basal MV2 medium with 0.2% FBS for 3 h and stimulated with VEGFA164 (50 ng ml−1, PeproTech) for 15 min. The activities of the Rho GTPases Rac1, CDC42 and RhoA were detected using kits from Cytoskeleton. Briefly, 300 µg of freshly prepared cell lysates were incubated with Rhotekin-RBD (RhoA) or PAK-PBD (CDC42, Rac1) beads for 1 h, 4 °C. Beads were washed five times and boiled in Laemmli buffer with β-mercaptoethanol. Samples were separated by SDS–PAGE (4–12% gradient gel) (Thermo Fisher Scientific), and blotting was performed to detect Rac1, CDC42 and RhoA with antibodies supplied in the kit.
Immunoprecipitation
Control or YES1-silenced HUVECs were starved at 37 °C in basal MV2 medium with 0.2% FBS for 3 h and stimulated with VEGFA164 (50 ng ml−1, PeproTech) for 15 min followed by lysis in 50 mM Tris-HCl pH 7.5, 5 mM EDTA, 150 mM NaCl and 0.5% NP-40) supplemented with protease and phosphatase inhibitor cocktails (Roche). Lysates (300 µg) were incubated with 1.5 µg VE-cadherin antibody (AF1002, R&D Systems) overnight at 4 °C. Protein G Sepharose beads (Cytiva) were incubated for 3 h at 4 °C. Beads were washed 5 times with lysis buffer, and proteins denatured and released from beads by boiling in β-mercaptoethanol-containing Laemmli buffer.
Western blot
Proteins were separated by SDS–PAGE (4–12% gradient gel) (Thermo Fisher Scientific), transferred to nitrocellulose membranes (GE Healthcare), and incubated sequentially with primary and horseradish peroxidase (HRP)-conjugated secondary antibodies. Signals were detected using enhanced chemiluminescence (Cytiva) and images retrieved using Bio-Rad ChemiDocMP and analyzed using Image Lab software (6.1). The following primary antibodies were used: rabbit anti-VE-cadherin Y685 (CP1981, ECM Biosciences, 1:1,000 dilution); goat anti-mouse VE-cadherin (AF1002, R&D Systems, 1:1,000 dilution); rabbit anti-pSrc (Y418) (44–660G, Thermo Fisher Scientific, 1:1,000 dilution); mouse anti-Yes (610376, BD Biosciences, 1:1,000 dilution); and mouse anti-p120-catenin (610133, BD Biosciences, 1:1,000 dilution); mouse anti-c-Src (05-184, Millipore, 1:1,000 dilution) and rabbit anti-GAPDH (2118, Cell Signaling, 1:5,000 dilution) The secondary antibodies used were HRP-conjugated anti-rabbit IgG (NA934, Cytiva), HRP-conjugated anti-mouse IgG (NA931, Cytiva) and HRP-conjugated anti-goat IgG (P0449, Dako). Secondary antibodies were used at a dilution of 1:7,500.
Statistics and reproducibility
Statistical analysis was performed using GraphPad Prism software (9.4.0). Statistical significance in comparisons between two groups was determined by two-tailed Student’s t-test or Welch’s t-test. Two-way ANOVA was used to compare between groups in time course experiments. Variances were similar between the groups compared. Differences were considered significant at P < 0.05. For animal experiments, no statistical methods were used to predetermine sample sizes. The experiments were not randomized. The investigators were not blinded to allocation during the experiment and outcome assessment.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Full source data for retinal EC distribution analysis is available on Zenodo (https://doi.org/10.5281/zenodo.7229061). Single-cell RNA-sequencing data generated from WT P6 and P10 retinas64 was obtained from the Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra), accession no. SRP322112. Additional data supporting the findings in this study are included in the main article and associated files. Source data are provided with this paper.
Code availability
Analysis code for retinal EC distribution can be accessed via https://github.com/wgiese/retina-vein-artery-cs
References
Claesson-Welsh, L., Dejana, E. & McDonald, D. M. Permeability of the endothelial barrier: identifying and reconciling controversies. Trends Mol. Med. 27, 314–331 (2021).
Komarova, Y. A., Kruse, K., Mehta, D. & Malik, A. B. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ. Res. 120, 179–206 (2017).
Wettschureck, N., Strilic, B. & Offermanns, S. Passing the vascular barrier: endothelial signaling processes controlling extravasation. Physiol. Rev. 99, 1467–1525 (2019).
Nitta, T. et al. Size-selective loosening of the blood–brain barrier in claudin-5-deficient mice. J. Cell Biol. 161, 653–660 (2003).
Honkura, N. et al. Intravital imaging-based analysis tools for vessel identification and assessment of concurrent dynamic vascular events. Nat. Commun. 9, 2746 (2018).
Richards, M. et al. Claudin5 protects the peripheral endothelial barrier in an organ and vessel-type-specific manner. eLife 11, e78517 (2022).
Corada, M. et al. Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl Acad. Sci. USA 96, 9815–9820 (1999).
Allingham, M. J., van Buul, J. D. & Burridge, K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J. Immunol. 179, 4053–4064 (2007).
Turowski, P. et al. Phosphorylation of vascular endothelial cadherin controls lymphocyte emigration. J. Cell Sci. 121, 29–37 (2008).
Schulte, D. et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 30, 4157–4170 (2011).
Wessel, F. et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol. 15, 223–230 (2014).
Hayer, A. et al. Engulfed cadherin fingers are polarized junctional structures between collectively migrating endothelial cells. Nat. Cell Biol. 18, 1311–1323 (2016).
Cao, J. et al. Polarized actin and VE-cadherin dynamics regulate junctional remodelling and cell migration during sprouting angiogenesis. Nat. Commun. 8, 2210 (2017).
Nanes, B. A. et al. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J. Cell Biol. 199, 365–380 (2012).
Adam, A. P. Regulation of endothelial adherens junctions by tyrosine phosphorylation. Mediators Inflamm. 2015, 272858 (2015).
Potter, M. D., Barbero, S. & Cheresh, D. A. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and β-catenin and maintains the cellular mesenchymal state. J. Biol. Chem. 280, 31906–31912 (2005).
Wallez, Y. et al. Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene 26, 1067–1077 (2007).
Orsenigo, F. et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat .Commun. 3, 1208 (2012).
Conway, D. E. et al. VE-cadherin phosphorylation regulates endothelial fluid shear stress responses through the polarity protein LGN. Curr. Biol. 27, 2727 (2017).
Li, X. et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 7, 11017 (2016).
Owen-Woods, C. et al. Local microvascular leakage promotes trafficking of activated neutrophils to remote organs. J. Clin. Invest. 130, 2301–2318 (2020).
Smith, R. O. et al. Vascular permeability in retinopathy is regulated by VEGFR2 Y949 signaling to VE-cadherin. eLife 9, e54056 (2020).
Arif, N. et al. PECAM-1 supports leukocyte diapedesis by tension-dependent dephosphorylation of VE-cadherin. EMBO J. 40, e106113 (2021).
Nawroth, R. et al. VE-PTP and VE-cadherin ectodomains interact to facilitate regulation of phosphorylation and cell contacts. EMBO J. 21, 4885–4895 (2002).
Grazia Lampugnani, M. et al. Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, β-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161, 793–804 (2003).
Juettner, V. V. et al. VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism. J. Cell Biol. 218, 1725–1742 (2019).
Reynolds, A. B. et al. SRChing for the substrates of Src. Oncogene 33, 4537–4547 (2014).
Schimmel, L. et al. c-Src controls stability of sprouting blood vessels in the developing retina independently of cell–cell adhesion through focal adhesion assembly. Development 147, dev185405 (2020).
Werdich, X. Q. & Penn, J. S. Src, Fyn and Yes play differential roles in VEGF-mediated endothelial cell events. Angiogenesis 8, 315–326 (2005).
Eliceiri, B. P. et al. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 4, 915–924 (1999).
Bernabeu, M. O. et al. PolNet: a tool to quantify network-level cell polarity and blood flow in vascular remodeling. Biophys. J. 114, 2052–2058 (2018).
Caolo, V. et al. Shear stress and VE-cadherin. Arterioscler .Thromb. Vasc. Biol. 38, 2174–2183 (2018).
Wang, Y. et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465, 483–486 (2010).
Drexler, H. C. A. et al. Vascular endothelial receptor tyrosine phosphatase: identification of novel substrates related to junctions and a ternary complex with EPHB4 and TIE2. Mol. Cell. Proteomics 18, 2058–2077 (2019).
Friedl, P. & Mayor, R. Tuning collective cell migration by cell–cell junction regulation. Cold Spring Harb. Perspect. Biol. 9, a029199 (2017).
Lee, H. W. et al. Role of venous endothelial cells in developmental and pathologic angiogenesis. Circulation 144, 1308–1322 (2021).
Fernandez-Chacon, M. et al. iSuRe-Cre is a genetic tool to reliably induce and report Cre-dependent genetic modifications. Nat. Commun. 10, 2262 (2019).
Barbacena, P. et al. Competition for endothelial cell polarity drives vascular morphogenesis in the mouse retina. Dev. Cell 57, 2321–2333 (2022).
Waschke, J., Burger, S., Curry, F. R., Drenckhahn, D. & Adamson, R. H. Activation of Rac-1 and Cdc42 stabilizes the microvascular endothelial barrier. Histochem. Cell Biol. 125, 397–406 (2006).
Amado-Azevedo, J. et al. A CDC42-centered signaling unit is a dominant positive regulator of endothelial integrity. Sci. Rep. 7, 10132 (2017).
Bentley, K. et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat. Cell Biol. 16, 309–321 (2014).
Thurston, G., Baldwin, A. L. & Wilson, L. M. Changes in endothelial actin cytoskeleton at leakage sites in the rat mesenteric microvasculature. Am. J. Physiol. 268, H316–H329 (1995).
Efimova, N. & Svitkina, T. M. Branched actin networks push against each other at adherens junctions to maintain cell–cell adhesion. J. Cell Biol. 217, 1827–1845 (2018).
Levinson, A. D., Oppermann, H., Levintow, L., Varmus, H. E. & Bishop, J. M. Evidence that the transforming gene of avian sarcoma virus encodes a protein kinase associated with a phosphoprotein. Cell 15, 561–572 (1978).
Erikson, R. L., Collett, M. S., Erikson, E. & Purchio, A. F. Evidence that the avian sarcoma virus transforming gene product is a cyclic AMP-independent protein kinase. Proc. Natl Acad. Sci. USA 76, 6260–6264 (1979).
Rohrschneider, L. R. Adhesion plaques of Rous sarcoma virus-transformed cells contain the src gene product. Proc. Natl Acad. Sci. USA 77, 3514–3518 (1980).
Azarnia, R., Reddy, S., Kmiecik, T. E., Shalloway, D. & Loewenstein, W. R. The cellular src gene product regulates junctional cell-to-cell communication. Science 239, 398–401 (1988).
Ferrando, I. M. et al. Identification of targets of c-Src tyrosine kinase by chemical complementation and phosphoproteomics. Mol. Cell Proteomics 11, 355–369 (2012).
Steed, E., Rodrigues, N. T., Balda, M. S. & Matter, K. Identification of MarvelD3 as a tight junction-associated transmembrane protein of the occludin family. BMC Cell Biol. 10, 95 (2009).
Martin, N. G., McAndrew, P. C., Eve, P. D. & Garrett, M. D. Phosphorylation of cyclin dependent kinase 4 on tyrosine 17 is mediated by Src family kinases. FEBS J. 275, 3099–3109 (2008).
Lee, S. W. et al. CD46 is phosphorylated at tyrosine 354 upon infection of epithelial cells by Neisseria gonorrhoeae. J. Cell Biol. 156, 951–957 (2002).
Varrin-Doyer, M. et al. Phosphorylation of collapsin response mediator protein 2 on Tyr-479 regulates CXCL12-induced T lymphocyte migration. J. Biol. Chem. 284, 13265–13276 (2009).
Takeda, H. et al. Comparative analysis of human SRC-family kinase substrate specificity in vitro. J. Proteome Res. 9, 5982–5993 (2010).
Tabula Muris, C. et al. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 562, 367–372 (2018).
Frye, M. et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J. Exp. Med. 212, 2267–2287 (2015).
Richards, M. et al. Intra-vessel heterogeneity establishes enhanced sites of macromolecular leakage downstream of laminin α5. Cell Rep. 35, 109268 (2021).
Schmidt, T. T. et al. Conditional deletion of FAK in mice endothelium disrupts lung vascular barrier function due to destabilization of RhoA and Rac1 activities. Am. J. Physiol. Lung Cell Mol. Physiol. 305, L291–L300 (2013).
Chen, X. L. et al. VEGF-induced vascular permeability is mediated by FAK. Dev. Cell 22, 146–157 (2012).
Kasahara, K. et al. Rapid trafficking of c-Src, a non-palmitoylated Src-family kinase, between the plasma membrane and late endosomes/lysosomes. Exp. Cell Res. 313, 2651–2666 (2007).
Sato, I. et al. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J. Cell Sci. 122, 965–975 (2009).
Hatanaka, K., Simons, M. & Murakami, M. Phosphorylation of VE-cadherin controls endothelial phenotypes via p120-catenin coupling and Rac1 activation. Am. J. Physiol. Heart Circ. Physiol. 300, H162–H172 (2011).
Grimsley-Myers, C. M. et al. VE-cadherin endocytosis controls vascular integrity and patterning during development. J. Cell Biol. 219, e201909081 (2020).
Neto, F. et al. YAP and TAZ regulate adherens junction dynamics and endothelial cell distribution during vascular development. eLife 7, e31037 (2018).
Zarkada, G. et al. Specialized endothelial tip cells guide neuroretina vascularization and blood–retina-barrier formation. Dev. Cell 56, 2237–2251 (2021).
Acknowledgements
We gratefully acknowledge E. Dejana (Uppsala University; IFOM Milano) and F. Osenigo (IFOM, Milano) for the pY658-VE-cadherin antibody. Euro-BioImaging ERIC and Biocenter Oulu Electron Microscopy core facility supported by University of Oulu and Biocenter Finland are acknowledged for providing infrastructure allowing ultrastructural analyses. We gratefully acknowledge A. Dubrac, Université de Montréal for providing annotations for the single-cell RNA-sequencing data of retinas, R. Benedito, Centro Nacional de Investigaciones Cardiovasculares for providing the iSuRe-Cre mouse model and R. Adams, Max-Planck Institute for providing Cdh5(PAC)-CreERT2 mice. This study was supported by the Swedish Research Council (2020-01349), the Knut and Alice Wallenberg foundation (KAW 2020.0057 and KAW 2019.0276), Fondation Leducq Transatlantic Network of Excellence Grant in Neurovascular Disease (17 CVD 03) to L.C.W., H.G., C.A.F. and M.B., Fundação para a Ciência e Tecnologia (PTDC/MED-PAT/31639/2017; CEECIND/04251/2017), European Research Council (679368) to C.A.F., Deutsche Forschungsgemeinschaft (KFO342, P2; CRC1450, B03) to D.V., the Academy of Finland (LE380986) and the Sigrid Jusélius Foundation to L.E. The processing of single-cell RNA-sequencing data was enabled by resources in project SNIC 2021/22-221 provided by the Swedish National Infrastructure for Computing at UPPMAX, partially funded by the Swedish Research Council through grant agreement no. 2018-05973.
Funding
Open access funding provided by Uppsala University.
Author information
Authors and Affiliations
Contributions
Y.J. designed the study, performed experiments and analyzed data. Y.D., M.R., M.K., A.S., L.S., E.B.A, A.P., E.N., M.J., M.S.J. and M.O.B. performed experiments, collected and analyzed data. W.G., E.B. and A.R. analyzed imaging data. S.N. analyzed single-cell RNA-sequencing data. K.H. characterized antibody specificity. M.O.B., M.W., E.G., C.A.F., D.V., L.E. and H.G. contributed to data analysis and interpretation. L.C.W. conceived, directed and financed the study. Y.J. and L.C.W. wrote the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Cardiovascular Research thanks Michael Simons and Gou Young Koh for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 pVE-cadherin antibody specificity and expression of YES1 and SRC.
(a) Validation of the specificity of the antibody against pY685 VE-cadherin by immunostaining in P6 retinas from WT (VEC-WT) and mutant VE-cadherin Y685F (VEC-Y685F) mice. A, artery; V, vein. (b) Immunostaining with antibody against pY731 VE-cadherin in retinas from WT and VE-cadherin Y731F mutant mice. (c) Immunostaining with pY731 VE-cadherin antibody in retinas from WT and VE-cadherin Y685F mutant mice. A, artery; V, vein. (d, e) Effectiveness of SRC or YES1 siRNA silencing in HUVECs 48 h after transfection shown by qPCR, n = 3 independent experiments. (f) HUVECs silenced for Yes (siYES1) cultured in static or flow (3 dyn/cm2) conditions and immunostained for pY658-VE-cadherin or total VE-cadherin protein. Scale bars, 50 µm. (g) Ratio of junctional pY658 area/total VE-cadherin area in the different conditions. n = 3 independent experiments. Note that quantification of pY658 signal was restricted to the junction area due to cross-reactivity with focal adhesions in vitro (see panel a) but not in vivo (see main Fig. 2a). (h-j) Relative expression levels of Yes1, Src and Cdh5 in different vessel types from single-cell sequencing analysis of P6 and P10 retinal ECs. Scale bars, 50 µm. Bar graphs show mean ± SEM with individual data points, two-tailed Student’s t-test.
Extended Data Fig. 2 Yes1 floxed mouse construction and immunostaining with pVE-cadherin antibodies.
(a) Schematic outline of targeting strategy to generate the Yes1 floxed model (Yes1 fl/fl, Cdh5CreERT2+; denoted Yes1 iECKO). (b) Yes1 and Src transcript levels in isolated lung endothelial cells from control (Yes1 fl/fl, Cdh5CreERT2-) or Yes1 iECKO mice at P6 shown by qPCR; tamoxifen treatment at P1-3. Control, n = 5; Yes iECKO, n = 8. (c-e) Immunofluorescence staining of Src iECKO P6 retinas showing total VE-cadherin (magenta) and phosphorylated VE-cadherin (green) pY685 (c), pY658 (d) and pY731 (e). Scale bars, 50 µm. Corresponding quantifications are shown in main Fig. 2g–i. (f) Immunostaining for pVE-cadherin (Y685) (green) and total VE-cadherin (magenta) in ECs of vena cava in P6 control and Yes1 iECKO mice. Scale bar, 20 µm. Quantification of normalized ratio of pVE-cadherin/total VE-cadherin of data in (f) is shown in (g), control, n = 5; Yes1 iECKO, n = 4. (h, i) Quantifications of pVE-cadherin immunostaining at Y685 and Y658 in P6 control and Yes1 iECHet retinas. Control, n = 3; Yes1 iECHet, n = 3. (j, k) Quantifications of pVE-cadherin immunostaining at Y685 and Y658 in P22 control and Yes1 iECKO retinas. Control, n = 3; Yes1 iECKO, n = 5. A, artery; V, vein. Bar graphs show mean ± SEM with individual data points, two-tailed Student’s t-test.
Extended Data Fig. 3 Silencing of PTPRB fails to rescue loss of pVE-cadherin by siYES1.
(a) Immunostaining of pY685 VE-Cad (green) and total VE-cad (magenta) in HUVECs with individual silencing of YES1, PTPRB or concomitant silencing of both. Scale bars, 50 µm. (b) Quantification of phospho/total VE-Cadherin in siControl, siYES1, siPTPRB and siYES1+siPTPRB HUVECs. n = 4 independent experiments. (c) Validation of PTPRB siRNA by qPCR. n = 3 independent experiments. Bar graph shows mean ± SEM with individual data points; two-tailed Student’s t-test.
Extended Data Fig. 4 Yes1 overexpression (iECOE), construction and characterization.
(a) Schematic outline of targeting strategy the of H11-CAG-STOP-Yes (Yes1 iECOE) mouse model. (b) Junctional localization of Yes in Yes1 iECOE retinas visualized by immunostaining of Yes (yellow) and VE-cadherin (magenta). Arrowheads indicate representative junctional localization of Yes. Scale bar, 10 µm. (c) Localization of Yes1 overexpressing ECs in vein (upper panel) and artery (lower panel) shown by immunostaining of Yes (red), pY685 VE-cadherin (green) and VE-cadherin (blue). Arrows indicate Yes1 overexpressing cells at branch points; arrowheads indicate control cells at branch points. Scale bars, 50 µm. (d) Normalized percentages of Yes1 OE cells and control cells at branch points in veins and arteries. n = 5 chimeric Yes1 iECOE retinas. Bar graph show mean ± SEM with individual data points; two-tailed Student’s t-test.
Extended Data Fig. 5 Yes and Src overexpression increase VE-Cad phosphorylation in vitro.
(a) Immunostaining of pY685 VE-Cadherin (green) and total VE-cadherin (magenta) in HUVECs transduced with mScarlet-tagged Yes or Src. Localization of Yes and Src proteins is indicated by the detection of mScarlet (gray). Boxed regions, shown enlarged in the lower right, highlight Yes-mScarlet localization at junctions and Src-mScarlet in the cytoplasm. Scale bars, 25 µm. The bright spots present in the mScarlet channel in YES1 but not SRC overexpressing cells were found reproducibly and could be a consequence of the degree of YES overexpression. (b) Quantification of phospho/total VE-Cad in control, Yes overexpression and Src overexpression HUVECs. n = 15 cells analyzed from 3 independent experiments for each group, Kruskal Wallis test. (c) Western blots showing the level of overexpression of Yes and Src protein. Quantifications are shown in (d, e), n = 3 independent experiments. Bar graph show mean ± SEM with individual data points; two-tailed Student’s t-test.
Extended Data Fig. 6 Enhanced migration of Yes-deficient ECs.
(a) Vasculature in P6 retinas of Cdh5 iECKO mice with chimeric recombination, shown by CD31 immunostaining (magenta). Distribution of non-recombined ECs with VE-cadherin expression detected by immunostaining (cyan). Dashed lines indicate an arterial region devoid of VE-cadherin+ ECs. V, vein; A, artery. Scale bar, 500 µm. (b) Individual HUVECs tracked manually during migration in a scratch wound assay. The tracks from cells at initial positions in rows 1, 2 or 3 are indicated by colors (first, red; second, yellow; third, green). Scale bars, 100 µm. (c) Migratory speed of individual cells calculated as track length (µm)/time (hour). Cells are grouped by their initial positions pooled from 3 independent experiments. siControl, n = 76, 59, 63 for cells initially in rows 1, 2, and 3, respectively; siYES1, n = 74, 58, 61 for cells initially in rows 1, 2, and 3, respectively. Mean values and individual data points are shown. (d) Migratory tracks of control and YES1-silenced HUVECs from (b) are shown with the initial position labeled as (0,0) for all cells irrespective of origin in first row; red, second row; yellow, third row; green. Dashed lines indicate the maximum traveled range of cells over 10 h. (e) Activation of CDC42, Rac1 and RhoA detected in control or YES1-silenced HUVECs with and without VEGFA treatment. Quantifications of activated CDC42, Rac1 and RhoA normalized to total protein levels (f-h). Bar graphs show mean ± SEM with individual data points; two-tailed Student’s t-test.
Extended Data Fig. 7 Vascular development in Yes1 iECKO, VE-cadherin Y685F mutant, Cdh5 iECKO and Yes1 iECOE mice.
(a) Retinal vascular development in Yes1 wt/wt, Cdh5CreERT2+ and Yes1 fl/fl, Cdh5CreERT2+ mice at P6. Scale bars, 500 µm. Quantification of the vascular area is shown in (b), Yes1 wt/wt, Cdh5CreERT2+, n = 6; Yes1 fl/fl, Cdh5CreERT2+, n = 12. (c) Filopodia at the sprouting front of P6 retinal vasculatures of control and Yes1 iECKO mice visualized by CD31 staining. Scale bars, 100 µm. (d) Quantification of the number of filopodia/100 µm sprouting front. Control, n = 6; Yes1 iECKO, n = 5. (e) CD31-positive retinal vasculature in VEC-WT and Y685F-VEC mice at P6. Scale bars, 500 µm. Quantification of the vascular area in littermates is shown in (f), WT, n = 9; Y685F, n = 9. (g) Vascular development in P6 retinas of control (Cdh5 wt/wt, Cdh5CreERT2+) and Cdh5 iECKO (Cdh5 fl/fl, Cdh5CreERT2+) mice, shown by CD31 immunostaining. Scale bars, 500 µm. (h) Quantification of the vascular area in control and Cdh5 iECKO retinas. Control, n = 6; Cdh5 iECKO, n = 4. (i) Retinal vasculature in H11-Yes1 0/0, Cdh5CreERT2+ and H11-Yes1 +/0, Cdh5CreERT2+ mice at P6. Scale bars, 500 µm. Quantification of the vascular area is shown in (j), H11-Yes1 0/0, n = 10; H11-Yes1 +/0, n = 10. Bar graphs show mean ± SEM with individual data points; two-tailed Student’s t-test.
Extended Data Fig. 8 Vascular sprouting, EC proliferation and apoptosis in Yes1 iECKO retinas.
(a) Aortic rings from the thoracic aorta from control or Yes1 iECKO mice cultured for 4 days with and without VEGFA. Vascular sprouts shown by CD31 immunostaining. Scale bars, 200 µm. Quantifications of the number and the length of the sprouts in different conditions shown in (b, c), control VEGFA-, n = 5; control VEGFA+, n = 4; Yes1 iECKO VEGFA-, n = 6; Yes1 iECKO VEGFA+, n = 6. Bar graphs show mean ± SEM with individual data points. (d) Endothelial cell proliferation in P6 retinas from control (Yes1 fl/fl, CDH5CreERT2-) or Yes1 iECKO mice shown by EdU staining (magenta) with ERG (green) co-staining to identify endothelial nuclei. Scale bars, 100 µm. Quantification shown in (e), control, n = 6; Yes1 iECKO, n = 5. (f) Apoptosis of endothelial cells in P6 retinas shown by co-staining of CD31 (magenta) and Cleaved Caspase3 (green). Arrows indicate Cleaved Caspase3+ ECs. Scale bars, 100 µm. Quantification is shown in (g), control, n = 4; Yes1 iECKO, n = 5. Bar graphs show mean ± SEM with individual data points; two-tailed Student’s t-test.
Extended Data Fig. 9 Adherens junction morphology in retinal vein and artery.
Junction morphology shown by immunostaining for VE-cadherin (magenta) and pY685 VE-cadherin (green) in a retinal artery (left) and vein (right) from P6 wt/wt mouse. Arrowheads indicate jagged junction morphology. Scale bar, 20 µm.
Extended Data Fig. 10 Shear stress and VEGFA-induced VE-cadherin internalization in YES1- and SRC-silenced HUVECs.
VE-cadherin internalization in control, Yes and Src silenced cells detected by antibody feeding assay in cultures exposed to flow (3 dyn/cm2) (a), or treated with VEGFA (50 ng/mL for 15 min, after the binding of anti-VE-cadherin antibody) (b). Immunofluorescent signals for internalized and total VE-cadherin shown in magenta and green respectively with nuclei (blue) co-stained with DAPI. Scale bars, 50 µm. Quantification shown in main Fig. 5d,e. (c) Immunoblotting using antibodies against p120-catenin and VE-cadherin on VE-cadherin immunoprecipitates (IP) from YES1-silenced HUVECs with or without VEGFA treatment (50 ng/mL for 15 min). GAPDH blotting for equal loading on total lysates run in parallel. (d) Quantification of p120 from the co-IP experiments, n = 3 independent experiments. Bar graphs show mean ± SEM with individual data points, two-tailed Student’s t-test was used for statistics.
Extended Data Fig. 11 Vascular integrity in Yes deficiency.
(a) Basal leakage of Evans blue in skin measured as Evans blue level = (Abs620-(abs500+abs740)/2)/weight. Control, n = 5; Yes1 iECKO, n = 7. (b, c) Blood-brain-barrier integrity assessed by measuring basal leakage of Evans blue (Control, n = 5; Yes1 iECKO, n = 7) and Cadaverine-Alexa-555 in mouse brains (Control, n = 6; Yes1 iECKO, n = 5). (d) pY685 VE-cadherin and VE-cadherin in adult mouse ear dermis vessels shown by immunofluorescence. V, vein; A, artery. Scale bars, 50 µm. Quantification shown in (e), control, n = 6; Yes1 iECKO, n = 7. Bar graphs show mean ± SEM with individual data points; two-tailed Student’s t-test.
Supplementary information
Supplementary information
Supplementary Table 1 and Supplementary Videos 1–12.
Supplementary Video 1
Live cell imaging of HUVECs transfected with control or YES1 siRNA in scratch wound healing assay. Images were taken every 15 min for 10 h. Migration of individual cells was tracked in ImageJ and their migratory routes are shown in color coded tracks (red, first row; yellow, second row, green, third row).
Supplementary Video 2
VE-cadherin dynamics shown by live imaging of HUVECs expressing GFP-tagged VE-cadherin and transfected with control siRNA. Images were taken every 52 s with confocal microscope. Examples of endocytosis of VE-cadherin vesicles are highlighted in boxed areas. Time stamps are min:s. Scale bar, 10 µm.
Supplementary Video 3
VE-cadherin dynamics shown by live imaging of HUVECs expressing GFP-tagged VE-cadherin and transfected with YES1 siRNA. Images were taken every 52 s with confocal microscope. Examples of the dynamics of VE-cadherin clusters are highlighted in boxed areas. Time stamps are min:s. Scale bar, 10 µm.
Supplementary Video 4
VE-cadherin dynamics shown by live imaging of HUVECs expressing GFP-tagged VE-cadherin and transfected with SRC siRNA. Images were taken every 15 s with confocal microscope. Examples of endocytosis of VE-cadherin vesicles are highlighted in boxed areas. Time stamps are min:s. Scale bar, 10 µm.
Supplementary Video 5
Live cell imaging of control and YES1-silenced HUVECs stained with SiR-actin dye. Images were taken every 1 min for 3 h. Scale bar, 50 µm.
Supplementary Video 6
Live cell imaging of migrating control HUVECs. Actin stress fibers visualized by SiR-actin staining. Time stamps are min:s. Scale bar, 10 µm.
Supplementary Video 7
Live cell imaging of migrating YES1-silenced HUVECs. Actin stress fibers visualized by SiR-actin staining. Arrows indicate the break of stress fibers. Time stamps are min:s. Scale bar, 10 µm.
Supplementary Video 8
Live cell imaging of SRC-silenced HUVECs during migration. Actin stress fibers visualized by SiR-actin staining. Time stamps are min:s. Scale bar, 10 µm.
Supplementary Video 9
Intravital imaging of VEGFA induced leakage in the dermal vessels in control mice. Images were taken every 2 s with confocal microscope and motion corrected in ImageJ. Scale bar, 50 µm.
Supplementary Video 10
Intravital imaging of VEGFA induced leakage in the dermal vessels in Yes1 iECKO mice. Images were taken every 2 s and motion corrected in ImageJ. Scale bar, 50 µm.
Supplementary Video 11
Video showing the appearance of individual leakage points in dermal vessels in control mice.
Supplementary Video 12
Video showing the appearance of individual leakage points in dermal vessels in Yes1 iECKO mice.
Source data
Source Data Fig. 1
Statistical Source Data for Fig. 1.
Source Data Fig. 1
Unprocessed Western Blots for Fig. 1.
Source Data Fig. 2
Statistical Source Data for Fig. 2.
Source Data Fig. 3
Statistical Source Data for Fig. 3.
Source Data Fig. 4
Statistical Source Data for Fig. 4.
Source Data Fig. 5
Statistical Source Data for Fig. 5.
Source Data Fig. 6
Statistical Source Data for Fig. 6
Source Data Fig. 7
Statistical Source Data for Fig. 7.
Source Data Extended Data Fig. 1
Statistical Source Data for Extended Data Fig. 1.
Source Data Extended Data Fig. 2
Statistical Source Data for Extended Data Fig. 2.
Source Data Extended Data Fig. 3
Statistical Source Data for Extended Data Fig. 3.
Source Data Extended Data Fig. 4
Statistical Source Data for Extended Data Fig. 4.
Source Data Extended Data Fig. 5
Statistical Source Data for Extended Data Fig. 5.
Source Data Extended Data Fig. 5
Unprocessed western blots for Extended Data Fig. 5.
Source Data Extended Data Fig. 6
Statistical Source Data for Extended Data Fig. 6.
Source Data Extended Data Fig. 6
Source Data Extended Data Fig. 6 Unprocessed western blots for Extended Data Fig. 6.
Source Data Extended Data Fig. 7
Statistical Source Data for Extended Data Fig. 7.
Source Data Extended Data Fig. 8
Statistical Source Data for Extended Data Fig. 8.
Source Data Extended Data Fig. 10
Statistical Source Data for Extended Data Fig. 10.
Source Data Extended Data Fig. 10
Unprocessed western blots for Extended Data Fig. 10.
Source Data Extended Data Fig. 11
Statistical Source Data for Extended Data Fig. 11.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jin, Y., Ding, Y., Richards, M. et al. Tyrosine-protein kinase Yes controls endothelial junctional plasticity and barrier integrity by regulating VE-cadherin phosphorylation and endocytosis. Nat Cardiovasc Res 1, 1156–1173 (2022). https://doi.org/10.1038/s44161-022-00172-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s44161-022-00172-z
This article is cited by
-
The BulkECexplorer compiles endothelial bulk transcriptomes to predict functional versus leaky transcription
Nature Cardiovascular Research (2024)
-
Report from the 2023 workshop on endothelial permeability, edema and inflammation
Nature Cardiovascular Research (2023)
-
YES to junctions, no to Src
Nature Cardiovascular Research (2022)
