
Abstract
The reprogramming of somatic cells to induced pluripotent stem cells (iPSCs) is lengthy and inefficient. The development of a reprogramming system that allows rapid and synchronous reprogramming to pluripotency is imperative for understanding the mechanism of iPSC formation and for future therapeutic applications. We have recently reported that a short expression in mouse primary B cells of the transcription factor C/EBPα before the induction of pluripotency factors increases the iPSC reprogramming efficiency >100-fold, involving 95% of the cells within a week. Here we present a dataset containing the time course of gene expression during this process as determined by microarray and RNA-seq techniques.
Design Type(s) | time series design • cell type comparison design |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | DNA microarray • next generation sequencing |
Factor Type(s) | Replicate • Timepoint • technology type • pulsed with C/EBPalpha |
Sample Characteristic(s) | Mus musculus • pre-B-lymphocyte • Embryonic stem cell • Induced pluripotent stem cell |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
Background and Summary
Reprogramming somatic cells into induced pluripotent cells (iPSCs) by Oct4, Sox2, Klf4 and Myc (OSKM)1 has revolutionized stem cell biology by opening unprecedented opportunities for cell therapy and disease modeling2. However, the finding that only a small proportion of the cells become reprogrammed, typically requiring >12 days, has hampered progress towards understanding the mechanism3. Recently, it has been proposed that reprogramming proceeds through a stochastic phase followed by a hierarchic phase in which the pluripotency network is established4–6. In addition, formation of iPSCs is preceded by the collapse of the somatic program and a mesenchymal to epithelial transition7–10, followed by the upregulation of endogenous pluripotency genes. Our earlier work showed that C/EBPα very efficiently and rapidly induces the transdifferentiation of immature and mature B cells into macrophages by upregulating macrophage genes and silencing the B cell program, without involving retrodifferentiation11–13. C/EBPα has also been reported to enhance the reprogramming efficiency of B cells to pluripotency when co-expressed with OSKM14,15, although the mechanism remains unknown and the final efficiency only reaches 1–3%.
We have recently found that an 18 h pulse of C/EBPα expression in B cells followed by OSKM activation induces a >100-fold increase in the iPSC reprogramming efficiency, involving up to 95% of the cells within a week16. Concomitantly, the cells undergo an epithelial to mesenchymal transition and pluripotency genes become upregulated to levels comparable to embryonic stem (ES) and iPS cells. In addition, B cells pulsed with C/EBPα (Bα′ cells) already initiate an epithelial to mesenchymal transition and downregulate the B cell program. In serum-free conditions, up to 70% Oct4-GFP positive cells are observed within 2 days, representing the fastest reprogramming to pluripotency reported so far. These results are consistent with the idea that the C/EBPα pulse helps to overcome the stochastic phase of iPSC reprogramming. The accelerated iPSC reprogramming approach described should help to fully elucidate the early events of reprogramming to pluripotency and, if applicable to human cells, could have potential clinical applications. Here, we describe gene expression profiling by two approaches, microarray and RNA-seq, during the reprogramming of mouse primary B cells, pulsed or not with C/EBPα, into iPSCs. Expression profiles from four time points across days 2 to 8 post-induction were analyzed in our previous work16. In this Data Descriptor we provide a detailed description of the full dataset, including the gene profiles from additional early time points not described earlier.
Methods
The following section is an expanded version of the methods description provided in Di Stefano et al.16
Overview
Mouse primary B cells were isolated from the Oct4-GFP reprogrammable mouse (containing a tetracycline-controlled transactivator (rtTa), a doxycycline-responsive OSKM cassette and an Oct4-GFP cassette)17 and infected with a C/EBPα-hCD4 retrovirus11. hCD4-positive B cells were sorted and incubated for 18 h with β-estradiol (E2) to activate C/EBPα, followed by inducer washout and OSKM activation by doxycycline treatment. The RNA was then collected at different time points during reprogramming (Fig. 1), including samples at 3, 6, 12 and 24 h post-induction (hpi) with OSKM that were not incorporated into our previous study16 but were actually part of the same experiment. In parallel, control B cells (not pulsed with C/EBPα) were exposed to doxycycline for OSKM induction and the RNA was collected following the scheme in Fig. 1. Embryonic stem cells and newly established iPSC lines (at passage 3) were used as a control for pluripotency gene expression.

Mouse primary B cells were purified from bone marrow of reprogrammable mice and infected with a retrovirus carrying an estradiol (E2) inducible form of C/EBPα. Infected cells were sorted based on the expression of the cell surface marker hCD4 and plated in B cell medium on OP9 stromal cells. B cells were then exposed for 18 h to E2, followed by washout of the inducer and addition of doxycycline to activate OSKM factors. Control B cells were directly induced with doxycycline. Cells were collected at the indicated time points and FACS sorted for live cells before RNA extraction. Gene expression profiling was performed by microarray and RNA-seq.
Mice
The reprogrammable mouse line containing a tetracycline-inducible OSKM cassette as well as rtTA has been described previously17. Twelve mice (B6 strain, males and females, all homozygous for the OSKM and rtTA cassettes) were used to collect the RNA samples.
Cell cultures and reprogramming
Isolation of hematopoietic cells from bone marrow was performed as previously described18. CD19+ cells (a mixture of pro-B and pre-B cells hereafter called B cells) were isolated using monoclonal antibody 1D3, purchased from BD Pharmingen, using MACS (Miltenyi Biotech). The purity of the sorted cell fractions (>98%) was confirmed by FACS using an LSR II flow cytometer (BD).
After isolation, B cells were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS) and IL-7 (10ng/ml) (B cell medium) for 1 week before inducing reprogramming on gelatinized plates seeded with a feeder layer of the OP9 stromal cell line.
Two different ESC lines (R1 (ATCC) and Bruce-4 (Millipore)) were used as positive controls for pluripotency gene expression. ESCs and B cell derived iPSCs were cultured on mitomycin C treated mouse embryo fibroblast (MEF) feeder cells in KO-DMEM medium (Invitrogen) supplemented with L-glutamine, penicillin/streptomycin, nonessential amino acids, β-mercaptoethanol, 1,000 U/ml LIF (ESC medium) and 15% FBS (Invitrogen) (ESC medium).
MEF cultures were established by trypsin digestion of mouse embryos (embryonic day 13.5) and the resulting cells cultured in DMEM supplemented with 10% FBS, L-glutamine and penicillin/streptomycin.
For the reprogramming, B cell medium was changed to ESC medium supplemented with 2 μg/ml of doxycycline and 15% FBS, IL-4 (10 ng/ml), IL-7 (10 ng/ml) and IL-15 (2 ng/ml). B cells and Bα′ cells were seeded at a density of 500 cells/cm2 in six-well plates. Bα′ cells were generated by exposing B cells for 18 h to 100 nM of E2 followed by inducer washout. To activate OSKM, the cells were subsequently treated with 2 μg/ml doxycycline. The reprogramming medium was changed every 2 days. For expansion of iPSC lines, colonies with ESC morphology were picked after doxycycline withdrawal at 12 days post induction and grown on MEF feeder layer in ESC medium.
Vectors and virus production and infection
The C/EBPαER-hCD4 retroviral vector has been described previously11 and was used to overexpress C/EBPα in B cells. The lentiviral vector expressing a constitutive dsRED has been generated by substituting the GFP with the dsRED gene in the PTYF vector19. This virus was utilized to generate αiPS cell lines constitutively expressing dsRED to follow chimera contribution upon injection into host blastocysts. Viral production was performed as reported earlier20. Briefly, HEK293T cells were co-transfected with the vector plasmid and packaging plasmids (p8.9 and VSVG) using calcium phosphate transfection. Viral supernatants were harvested 48–72 h later and concentrated by ultracentrifugation at 20,000 g for 2 h at 20 °C. Viral concentrates were re-suspended in 1× PBS and stored at −80 °C. Infection of B cells was carried out by centrifugation of the cells with concentrated virus for 2 h at 32 °C at 1000 g in B cell medium.
Chimeric mice
For the chimera formation assay, 10 to 15 iPSCs were injected into a 3.5 days post coitum (dpc) blastocyst of CD1 mice and transferred into pseudo-pregnant 2.5 dpc recipients for chimera generation. Chimerism was ascertained by the expression of the lentiviral PGK-dsRED.
RNA isolation and quantification
RNA isolation from MEFs and B cells was performed with the miRNeasy Mini Kit (Qiagen). RNA was collected from sorted live cells at 0, 3, 6, 12, 24, 48, 96, 144 and 192 hpi. To remove the feeders, ESCs and iPSC clones at passage 2 or higher were seeded on gelatinized plates and processed with the above kits. RNA was eluted from the columns using RNase-free water or TE buffer and quantified by Nanodrop.
Gene expression arrays
RNA samples (with an RNA integrity number (RIN) >9) were subjected to gene expression analyses using Agilent expression arrays. Briefly: 100 ng of total RNA was labeled using LowInputQuick Amp Labeling Kit (Agilent 5190-2305) following the manufacturer’s instructions. mRNA was reverse transcribed in the presence of T7-oligo-dT primer to produce cDNA. cDNA was then in vitro transcribed with T7 RNA polymerase in the presence of Cy3-CTP to produce labeled cRNA. The labeled cRNA was hybridized to the Agilent SurePrint G3 gene expression 8×60 K microarray according to the manufacturer’s protocol. The arrays were washed and scanned on an Agilent G2565CA microarray scanner at 100% PMT and 3 μm resolution. Intensity data were extracted using the Feature Extraction software (Agilent). Raw data were taken from the Feature Extraction output files and corrected for background noise using the normexp method21. To assure comparability across samples, we used quantile normalization. Probe Level Quantile Normalization of High Density Oligonucleotide Array Data. http://bmbolstad.com/stuff/qnorm.pdf). (Bolstad, B. (2001)).
RNA-seq
Libraries were prepared using the TruSeq Stranded mRNA Sample Prep Kit v2 (ref. RS-122-2101/2, Illumina) according to the manufacturer’s protocol. Briefly, 900 ng of total RNA was used for poly(A)-mRNA selection using streptavidin-coated magnetic beads, and subsequently fragmented to approximately 300 bp. cDNA was synthesized using reverse transcriptase (SuperScript II, ref. 18064–014, Invitrogen) and random primers. The second strand of the cDNA incorporated dUTP in place of dTTP. Double-stranded DNA was further used for library preparation. DNA was subjected to A-tailing and ligation of the barcoded Truseq adapters. All purification steps were performed using Qiagen PCR purification columns (refs. 50928106 and 50928006). Library size-selection was done with 2% low-range agarose gels. Fragments with insert sizes of 200 to 400 bp were cut out from the gel, and DNA was extracted using QIAquick Gel Extraction Kit (ref. 50928706, Qiagen) and eluted in 20 μl EB. Library amplification was performed by PCR on the size-selected fragments using the primer cocktail supplied in the kit.
Finally, the libraries were analyzed using Agilent DNA 1000 chips to determine the quantity and size distribution, and then quantified by qPCR using the KAPA Library Quantification Kit (ref. KK4835, KapaBiosystems) prior to amplification with Illumina’s cBot. Libraries were loaded at a concentration of 10 pM onto the flowcell and sequenced on Illumina’s HiSeq 2000. Base-calls were performed with CASAVA-1.8.2 software (Illumina). Reads were aligned to the mm9 genome with STAR v2.3.0.1, keeping only uniquely mapped reads with a maximum of two mismatches (option: outFilterMismatchNmax 2, outFilterMultimapNmax 1). Gene expression quantification was performed with the software HTSeq-count (option: mode union, stranded, features exons, attribute gene_id) on the RefSeq mm9 annotation (from the UCSC Genome Browser; the exact version used here is available on demand). Genes with uncounted reads in both conditions were removed, and normalization was performed with DESeq (option: method blind, sharingMode fit-only, fitType local).
Data Records
Gene expression profiling was performed, using both microarrays and RNA-seq approaches, on RNA samples collected from cells at 0, 3, 6, 12, 24, 48, 96, 144 and 192 hpi. Two biological replicates are available for each time point. Data from time points 0, 48, 96, 144 and 192 were analyzed in our previous work16. The full dataset is available in association with this Data Descriptor, including data from four earlier time points not analyzed in the previous work. A quality control report from Agilent Features Extraction software is also included. All samples are summarized in Table 1.
Data record 1
GEO accession number GSE46321 (Data Citation 1) contains microarray data for all samples listed in Table 1.
Data record 2
RNA-seq data for the B cell (T0_1) and Bα′ cell (aT0_1) samples are available under the GEO accession number GSE52396 (Data Citation 2).
Data record 3
The normalized microarray data for all the samples are available in Figshare (Data Citation 3).
Data record 4
A Quality Control report from the Feature Extraction software for the microarray data is available in Figshare (Data Citation 2).
Technical Validation
Confirmation of pluripotency
Bone marrow isolated mouse primary B cells were pulsed with C/EBPα for 18 h and doxycycline treated to activate the OSKM cassette in ES medium on inactivated OP9 feeder cells. After doxycycline washout at day 12, transgene independent Oct4-GFP clones were picked and expanded in ES medium as stable iPSC lines (referred to as αiPSCs) (Fig. 2a). In order to assess the in vivo differentiation potential of the expanded cell lines, iPSCs expressing dsRED and Oct4-GFP were injected into host blastocysts. High contribution to chimerism was observed in developing embryos (Fig. 2b).

(a) Oct4-GFP+ αiPS cells generated from mouse primary B cells expressing dsRED under the constitutive PGK promoter. (b) Contribution of aiPS#3 dsRED-labeled cells to chimera formation after blastocyst injection. White arrow indicates a dsRED-expressing chimeric mouse.
Quality control of high throughput data
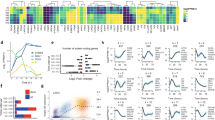
For microarrays (Data Records 1, 3 and 4; Data Citations 1 and 3), after background correction and quantile normalization, we looked at sample correlation at the probe level (Fig. 3a). All pairs of replicates correlate very well (the diagonal of the heatmap), and all pluripotent stem cell samples show a very high correlation (lower right block). Principal component analysis (PCA) (Fig. 3b) confirmed the similarity between replicates and all pluripotent stem cells (ES/iPS), the absence of major changes during OSKM-only induction (green points) and the trajectory from B cells to pluripotent cells in Bα′+OSKM cells. The high correlation between all the OSKM-only samples (T0 to T192) was further confirmed by qRT-PCR analysis16, showing that gene expression in these remains essentially unchanged.

(a) Heatmap showing Pearson correlation for duplicate microarray samples; the lower limit is 0.8 because correlation was calculated on all probes and most of them do not exhibit any change under any condition, leading to a high correlation between all samples. (b) Principal component analysis. Percentage of total variance explained by PC-1: 36%; by PC-2: 21%. (c) Distribution of RNA-seq sequencing quality along the reads, for B cells (left) and Bα’ cells (right). The X-axis corresponds to the position in the read and the Y-axis reflects the sequencing quality, with box plots representing 10th–25th–75th and 90th percentiles over all the reads. The lower part of the Y-axis scale has been omitted because all the observed values are above 30. The similarities between the B and Bα’ samples indicate uniformly high quality. For more explanations see technical validation part.
For RNA-seq data (Data Record 2; Data Citation 2), we sequenced 87,105,048 and 83,487,348 reads for B and Bα′ cells, respectively. Sequencing quality was good and did not indicate read bias (Fig. 3c). Using the STAR aligner22, we found 69,716,252 and 70,864,793 reads uniquely mappable onto the mm9 genome, and found 52,070,014 and 55,749,585 reads (60 and 67% of total sequenced reads) mapping to an annotated exon of the RefSeq mm9 annotation, indicating high quality mRNA sequencing.
Usage Notes
For gene expression microarray data, differentially expressed genes can be identified using various methods, in particular the popular LIMMA R package23 or the EDGE software24. For RNA-seq, we recommend using TopHat as a read aligner. Recent protocols explain how to process and analyze RNA-seq data25,26.
Additional information
How to cite this article: Di Stefano, B. et al. Time-resolved gene expression profiling during reprogramming of C/EBPα-pulsed B cells into iPS cells. Sci. Data 1:140008 doi: 10.1038/sdata.2014.8 (2014).
References
References
Takahashi, K., Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676 (2006).
Robinton, D. A., Daley, G. Q. The promise of induced pluripotent stem cells in research and therapy. Nature 481, 295–305 (2012).
Stadtfeld, M., Hochedlinger, K. Induced pluripotency: history, mechanisms, and applications. Genes Dev. 24, 2239–2263 (2010).
Hanna, J. H., Saha, K., Jaenisch, R. Pluripotency and cellular reprogramming: facts, hypotheses, unresolved issues. Cell 143, 508–525 (2010).
Buganim, Y. et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150, 1209–1222 (2012).
Buganim, Y., Faddah, D. A., Jaenisch, R. Mechanisms and models of somatic cell reprogramming. Nat. Rev. Genet. 14, 427–439 (2013).
Samavarchi-Tehrani, P. et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 7, 64–77 (2010).
Li, R. et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 7, 51–63 (2010).
Hansson, J. et al. Highly coordinated proteome dynamics during reprogramming of somatic cells to pluripotency. Cell Reps 2, 1579–1592 (2012).
Polo, J. M. et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151, 1617–1632 (2012).
Xie, H., Ye, M., Feng, R., Graf, T. Stepwise reprogramming of B cells into macrophages. Cell 117, 663–676 (2004).
Bussmann, L. H. et al. A robust and highly efficient immune cell reprogramming system. Cell Stem Cell 5, 554–566 (2009).
Di Tullio, A. et al. CCAAT/enhancer binding protein alpha (C/EBP(alpha))-induced transdifferentiation of pre-B cells into macrophages involves no overt retrodifferentiation. Proc. Natl Acad. Sci. USA 108, 17016–17021 (2011).
Hanna, J. et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 133, 250–264 (2008).
Eminli, S. et al. Differentiation stage determines potential of hematopoietic cells for reprogramming into induced pluripotent stem cells. Nat. Genet. 41, 968–976 (2009).
Di Stefano, B. et al. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature 506, 235–239 (2014).
Carey, B. W., Markoulaki, S., Beard, C., Hanna, J., Jaenisch, R. Single-gene transgenic mouse strains for reprogramming adult somatic cells. Nat. Methods 7, 56–59 (2010).
Lo Celso, C., Scadden, D. Isolation and transplantation of hematopoietic stem cells (HSCs). J. Vis. Exp 2, 157 (2007).
He, J., Kallin, E. M., Tsukada, Y., Zhang, Y. The H3K36 demethylase Jhdm1b/Kdm2b regulates cell proliferation and senescence through p15(Ink4b). Nat. Struct. Mol. Biol. 15, 1169–1175 (2008).
Di Stefano, B. et al. A microRNA-based system for selecting and maintaining the pluripotent state in human induced pluripotent stem cells. Stem Cells 29, 1684–1695 (2011).
Ritchie, M. E. et al. A comparison of background correction methods for two-colour microarrays. Bioinformatics 23, 2700–2707 (2007).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Molec. Biol. 3, 3 (2004).
Storey, J. D., Xiao, W., Leek, J. T., Tompkins, R. G., Davis, R. W. Significance analysis of time course microarray experiments. Proc. Natl Acad. Sci. U.S.A. 102, 12837–12842 (2005).
Anders, S. et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786 (2013).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Data Citations
Di Stefano, B., Collombet, S., & Graf, T. Gene Expression Omnibus GSE46321 (2014)
Di Stefano, B., Collombet, S., & Graf, T. Gene Expression Omnibus GSE52396 (2014)
Di Stefano, B., Collombet, S., & Graf, T. Figshare https://doi.org/10.6084/m9.figshare.939408 (2014)
Acknowledgements
We would like to thank Francesco Limone for his help with the figures, Laura Batlle for the generation of chimeric mice and the CRG genomic facility for high throughput sequencing and help with the bioinformatics analysis.
Author information
Authors and Affiliations
Contributions
T.G. and B.D.S. designed the project and the experiments and wrote the paper. B.D.S. performed the cell culture, the reprogramming experiments, the RNA collections and the molecular biology. S.C. was in charge of the bioinformatics analysis. This work was supported by the Ministerio de Educación y Ciencia, SAF.2007-63058 and AGAUR 2009 SGR768. B.D.S. holds a La Caixa International PhD Fellowship.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc/3.0/ Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Di Stefano, B., Collombet, S. & Graf, T. Time-resolved gene expression profiling during reprogramming of C/EBPα-pulsed B cells into iPS cells. Sci Data 1, 140008 (2014). https://doi.org/10.1038/sdata.2014.8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2014.8
