
Abstract
Recent evidence suggests that experimental induction of hepatocytes into pancreatic cells provides new cell transplantation therapy prospects for type 1 diabetes mellitus. Stepwise differentiation from rat liver epithelial stem-like WB-F344 cells (WB cells) into functional insulin-secreting cells will identify key steps in β-cell development and may yet prove useful for transplantation therapy for diabetic patients. An essential step in this protocol was the generation of pancreatic precursor cell that express Pdx1 based on induction by a combination of 5-aza-2′-deoxycytidine, trichostatin A, retinoic acid and a mix of insulin, transferrin and selenite. The Pdx1-expressing cells express other pancreatic markers and contribute to endocrine cells in vitro and in vivo. This study indicates an efficient chemical protocol for differentiating WB cells into functional insulin-producing cells using small molecules and represents a promising hepatocyte-based treatment for diabetes mellitus.
Similar content being viewed by others
Introduction
A progressive decrease in the pancreatic β-cell mass is the main cause of diabetes and can be counterbalanced by providing new insulin-producing cells (IPCs). Promising islet cell transplantation methods as a means of reversing type 1 diabetes mellitus (DM) have been hampered by islet availability as well as by allograft rejection1,2. Liver-to-endocrine pancreas transdifferentiation is an attractive strategy for generating β-cell surrogates since the liver and pancreas share a common bipotential precursor cell within the embryonic endoderm3,4. Most previously reported induction strategies required genetic manipulation and induced cells were incomplete and non-selective for β-cell phenotypes and function5,6,7.
Recent progress has been made demonstrating that some small molecules can promote definitive cellular differentiation to induce pluripotent stem cells (PSCs)6,8 and modulate various stages of β-cell differentiation from induced PSCs9,10. For example, 5-aza-2′-deoxycytidine (5-AZA), an inhibitor of DNA methylase, was used to successfully induce Ngn3 expression and endocrine differentiation in the PANC-1 human ductal cell line11. Trichostatin A (TSA), a regulator of chromatin remodeling, triggers the process of dedifferentiation and further endocrine pancreatic differentiation12,13. Retinoic acid (RA) facilitates the development of Pdx1 pancreatic endocrine progenitor cells and their further differentiation into β-cells14,15. Nicotinamide promotes transdifferentiation and maturation of liver stem cells into IPCs16.
There have already been several reports on the generation of IPCs using small molecules from various sources of stem cells, such as embryonic stem cells and mesenchymal stem cells. However, to date, there has been no report demonstrating that pancreatic β-cells can be induced from liver stem cells in this way. Therefore, by mimicking embryonic pancreatic development, we explored a stepwise protocol for the formation of IPCs from WB cells to control differentiation in vitro. In this study, efforts were focused on the key step of generating Pdx1-expressing cells as pancreatic progenitors. Several methods for inducing differentiation from stem cells into Pdx1-positive cells have been recently reported2,14,17,18, but the combination and sequence of small molecules for induction and differentiation remain to be optimized.
We present here a three-step approach for generating functional IPCs from WB cells within only 17 days. A sequential treatment with 5-AZA, TSA, RA and insulin, transferrin and selenite (ITS) induced efficient differentiation of WB cells into pancreatic precursor cells (WB-A cells). More than 5% of the cells became Pdx1-expressing pancreatic progenitors in only 10 days, eventually becoming IPCs in a step requiring nicotinamide for 7 days in vitro or transplantation into diabetic nude rats for 60 days in vivo. These IPCs expressed characteristic pancreatic β-cell marker genes, such as insulin I, insulin II, GK with no evidence of non-β-gene expression. Moreover, insulin was released in response to glucose concentration and the induced IPCs in vivo could ameliorate hyperglycemia in diabetic rats. The identification of an attractive chemical strategy that induces hepatocyte-derived IPCs is an important approach for diabetes cell therapy in the near future.
Results
Reprogramming of WB cells into Pdx1-positive cells
A stepwise protocol for screening combinations of small molecules was designed to lead to the formation of IPCs from WB cells (Fig. 1a). First, we focused on generating Pdx1-expressing cells before inducing IPCs. The empirical approach involved selecting induction factors such as 5-AZA, TSA, ITS and RA and testing them using various concentrations and application methods, as well as assorted medium formulations. In the first stage, cells were cultured for 3 days in the presence of 5 μM 5-AZA for 2 days and 100 nM TSA for 1 day. We adopted 5 μM as the optimal concentration of 5-AZA by testing cell survivability after increasing doses of 1–5 μM. Concentrations of more than 5 μM resulted in increased cell death and reduced differentiation capability (data not shown). Parental WB cells were initially small and polygonal in shape (Fig. 1b). During the first stage, the rate of cell proliferation decreased with metamorphosis into spindle-shaped cells. To determine whether these morphological changes reflected successful dedifferentiation of WB cells, we used both semi-quantitative and quantitative reverse transcription polymerase chain reaction (RT-PCR) to analyze the specific gene expression patterns for the hepatocyte dedifferentiation marker enhancer C/EBPβ and liver genes for ALB and AFP19,20,21. Expression of the C/EBPβ gene was drastically downregulated, while AFP and ALB were undetectable after stage 1 (Fig. 2a.i and Fig. 2b.i). In the next stage, after exposure to 100 nM TSA, cells were supplemented with a serum-free medium containing 1×ITS and 2 μM RA for 7 days (Fig. 1a). The cells continued to differentiate to form smaller cells with a higher nucleus/cytoplasm ratio than WB cells. To determine whether these morphological changes reflected successful differentiation of WB cells into pancreatic precursor cells, we analyzed specific gene expression patterns of the pancreatic progenitor marker Pdx1 by RT-PCR after step 2. As shown in Fig. 2a.ii and Fig. 2b.ii, the Pdx1 gene was activated. These results showed that efficient conversion of WB cells into Pdx1-expressing progenitor cells occurred after step 2.

Differentiation of WB cells into IPCs by a stepwise protocol and stage-specific cell morphology.
(a) Schematic representation of the three-step protocol to derive IPCs from WB cells. (b) Stage-specific cell morphology. Scale bar: 100 μm.

Gene expression analysis using semi-quantitative RT-PCR and quantitative RT-PCR.
Rat liver served as a positive control to estimate expression levels of the dedifferentiation of WB cells. Rat pancreas served as a positive control to estimate expression levels achieved in the differentiated WB-A cells. (a) Gene expression analysis using semi-quantitative RT-PCR. (a.i) Expression of genes related to liver markers and hepatocyte dedifferentiation marker. (a.ii) Expression of genes related to β-cell development. (a.iii) Expression of genes related to β-cell function. (a.iv) Expression of genes related to endocrine and exocrine markers. (b) Gene expression analysis using quantitative RT-PCR. mRNA of liver as a control to normalize data in (b.i). mRNA of WB cells as a control to normalize data in (b.ii) to (b.iv). (b.i) Expression of genes related to liver markers and hepatocyte dedifferentiation marker. (b.ii) Expression of genes related to β-cell development. (b.iii)Expression of genes related to β-cell function. (b.iv) Expression of genes related to endocrine and exocrine markers.
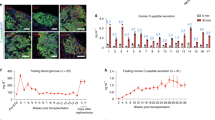
To determine whether the newly generated WB-A cells had undergone pancreatic differentiation, we detected the expression of various genes related to pancreas development and β-cell function by RT-PCR and immunofluorescence (Fig. 2a.ii-iv, Fig. 2b.ii-iv and Fig. 3a, b). At day 10 (the end of step 2), compared to WB cells, the WB-A cells expressed multiple genes characteristic of the key pancreatic early-stage developmental factors including Pdx1, Ngn3, NKX2.2 and the endocrine factor insulin I in WB-A cells. However, expression of late-stage developmental genes Pax4, Pax6 and MafA was not detected. Although functional β-cell genes including GLUT2, PC1/3 and PC2 were positive in WB-A cells, it is insignificant when compared to WB cells (Fig. 2a.ii–iv and Fig. 2b.ii–iv). These results suggest that only early differentiation to pancreatic cells occurred. Non-β-cell endocrine markers, such as somatostatin, glucagon, pancreatic polypeptide (PP) and exocrine marker amylase, were not detected (Fig. 2a.iv and Fig. 2b.iv), suggesting only β-cell differentiation occurred with this protocol. Although the induced WB-A cells expressed the pancreatic endocrine gene for insulin I (Fig. 2a.iv, Fig. 2b.iv and Fig. 3b), they were not capable of glucose-responsive insulin release (Fig. 3c). Taken together, the results suggest that these conditions supported the early differentiation of pancreatic cells without mature β-cell function after step 2.

Pancreatic gene expression in WB-A cells detected by immunofluorescence and insulin release analysis.
INS-1 and WB cells served as positive and negative controls, respectively. (a) Immunofluorescence analysis of Pdx1 expression was positive in WB-A cells in stage 2. Pdx1 (red), Hoechst-33342 for nuclear (blue). (b) Immunofluorescence analysis of insulin expression was positive in WB-A cells. Insulin (green). (c) Insulin release was not responsive to glucose stimulation in WB-A cells. Scale bar: 100 μm. **P < 0.01.
To confirm the efficiency of inducing Pdx1-positive cells, we observed that over 5% of all WB cells within a population definitively differentiated into Pdx1-expressing cells, as confirmed by immunofluorescence with the Pdx1 antibody (Fig. 3a). This was higher than the less than 1% previously reported for cells analyzed at the same time, induced by the exogenous transduction gene22. Thus, the combination of 5-AZA, TSA, RA and ITS efficiently induced definite pancreatic endoderm formation of Pdx1-expressing cells derived from WB cells.
Further differentiation into IPCs from pancreatic precursor cells in vitro
To further promote the differentiation of WB-A cells into IPCs, the differentiated cells from stage 2 were transferred to a culture medium supplemented with nicotinamide for about 7 days, which promotes the maturation of pancreatic β-cells16. The cells differentiated to a decreased nucleus/cytoplasm ratio with an increased cytoplasm but did not form aggregates (Fig. 1b). To detect whether endoderm- and pancreatic-specific genes were expressed in the differentiated cells, RT-PCR and immunofluorescence staining were performed at each stage of the differentiation process (Fig. 2a.ii–iv and Fig. 2b.ii–iv and Fig. 3a, b and Fig. 4a, b). At the end of stage 3, the islet cell-specific marker genes Pdx1, NeuroD and insulin I were also expressed as in stage 2, whereas the Pax4, Pax6 and MafA genes, the β-cell functional GK, Kir6.2 and the endocrine insulin II gene were activated (Fig. 2a.ii–iv and Fig. 2b.ii–iv). The expression of PC1/3, PC2 and GLUT2 was upregulated than that in stage 2 (Fig. 2a.iii and Fig. 2b.iii). We still did not detect somatostatin, glucagon and PP (Fig. 2a.iv and Fig. 2b.iv). The expression of ALB, AFP and C/EBPβ was undetectable in stage 2 and 3 (Fig. 2a.i and Fig. 2b.i ). Furthermore, immunofluorescence staining showed that insulin was positive in near 10% Pdx1-expressing cells (insulin was detected in 0.5% cells of the whole population) (Fig. 4a, b and Supplementary Fig. 1). To test the functional ability of IPCs, we examined the glucose dependency of insulin secretion after treatment with 5.5 mM or 25 mM glucose. The insulin secretion of IPCs was 1.068 ± 0.081 mU/L per 105 cells after exposure to 5.5 mM glucose and increased to 3.199 ± 0.226 mU/L per 105 cells after exposure to 25 mM glucose (Fig. 4c). Taken together, these data provide the specific expression pattern of these important pancreatic β-cell hormones and transcription factors and indicate selective transdifferentiation into islet-like β cells at the final stage in vitro. These results indicate that the WB-A cells have the potential to differentiate into pancreatic hormone-expressing cells.

WB-A cells formed IPCs in vitro.
INS-1 and WB cells served as positive and negative controls, respectively. (a) Immunofluorescence analysis of Pdx1 expression was positive in IPCs in stage 3. Scale bar: 100 μm. (b) Immunofluorescence analysis of insulin expression was positive in IPCs in stage 3. Scale bar: 100 μm. (c) IPCs in stage 3 released insulin upon glucose stimulation. **P < 0.01. (d) TEM image of induced IPCs in stage 3. IPCs showed secretary vesicles (black arrows). Scale bar: 500 nm.
At the ultrastructural level, induced IPCs showed scattered cytoplasmic globular structures containing insulin molecules that were similar to those seen in β-cells and confrmed by transmission electron microscopy (TEM) (Fig. 4d). These results indicate that induced cells could indeed process insulin and form insulin secretory granules.
WB-A cells can form IPCs in vivo
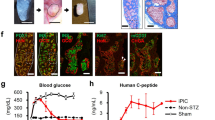
To further assess the developmental competence of WB-A cells and determine if they possessed the ability to further differentiate into functional pancreatic endocrine β-like cells, we transplanted the WB-A cells into the left renal capsules of streptozotocin-treated (STZ-treated) diabetic nude rats (n = 6), observed blood glucose and insulin changes during transplantation (Tx). As demonstrated in Fig. 5a, rats receiving the transplant began to have normalized blood glucose levels (less than 13.9 mM) within 15 days. Furthermore, upon explantation (Ex) of the implanted WB-A cells, around day 36, persistent hyperglycemia returned, while animals that retained the transplanted cells maintained fairly normal glucose levels, confirming that the implanted WB-A cells were indeed responsible for the reduction in blood glucose levels. In contrast, animals that received phosphate-buffered saline (PBS) sham-operation or received WB cells exhibited persistent hyperglycemia and died between days 32 and 39 because they did not receive insulin treatment. Fasting serum rat insulin levels were also increased to 2 fold in WB-A Tx rats at day 30 after Tx compared to those at day 3 before Tx (Fig. 5b). However, the transplanted cells did not show proliferation in vivo, which was revealed by the negative immunostaining of Ki67 protein antigen in the kidney paraffinn-embedded sections at day 60 post-transplantation (data not show). Meanwhile no tumors were detected in any of the rats transplanted with WB or WB-A cells.

WB-A cells formed IPCs in vivo.
(a) Blood glucose levels were monitored regularly under fasting conditions in WB-A Tx rats (n = 6).WB Tx rats (n = 6) as controls and PBS Tx rats (n = 3) as negative controls. Arrow: Ex. **P < 0.01 (b) Insulin levels were monitored under fasting conditions pre and post Tx. **P < 0.01 (c) Results of IPGTT with WB-A Tx DM rats (n = 3). DM rats (n = 3) and normal rats (n = 5) served as positive and negative controls respectively. (d) Results of glucose-stimulated insulin release assay with WB-A Tx DM rats. (e) HE and immunofluorescence analysis of insulin in the explanted WB-A cells. Rat pancreas served as positive controls. Sections were immunostained with antibodies to insulin (1:500). Scale bar: 100 μm.
To further determine the ability of the transplanted, liver-derived IPCs to respond to a glucose challenge, we performed intraperitoneal glucose tolerance test (IPGTT) and glucose-stimulated insulin release assay on the transplanted WB-A cell rats (n = 3) and on positive controls (DM) rats (n = 3) and negative controls (normal) rats (n = 5) after 60 days post-transplantation. As illustrated in Fig. 5c and Fig. 5d, rats receiving WB-A cells had a similar pattern to normal control rats without cell transplantation. These data suggest that these liver-derived IPCs indeed further differentiated and matured into functional pancreatic β-like cells.
To further characterize the identity of the transplanted WB-A cells, we examined the histological characteristics and insulin protein expression in the explanted cells after in vivo differentiation at day 60 post-transplantation. The explanted WB-A cells formed glandular structure-like β-cells using hematoxylin and eosin (HE) (Fig. 5e). Immunofluorescence staining for insulin protein showed that some of the implanted cells expressed insulin (Fig. 5e). These findings suggest that the WB-A cells had further differentiated into mature functional IPCs in a high-glucose microenvironment in vivo with the ability to ameliorate hyperglycemia by secreting insulin, like pancreatic β-cells.
Gene-expression profiles of pre- and post-transplanted WB-A cells
To explore the underlying molecular mechanism responsible for the functional shift of WB-A cells in vivo, we compared the gene expression profiles of post-transplanted WB-A cells at day 60 with IPCs at stage 3 and WB-A cells at stage 2 as well as with their parental WB cells. Several noticeable changes were seen in the expression of some genes related to β-cell development and function ((Fig. 2a.ii–iv and Fig. 2b.ii–iv)). First, after 60 days in vivo, the transplanted cells expressed the PC1, PC2, Pax4, Pax6, MafA, insulin II, GK and Kir6.2 genes, which were not expressed in WB-A cells and which were upregulated compared with IPCs in vitro. Second, Ngn3 was downregulated more than in IPCs in vitro and WB-A cells. Third, Pdx1 was also expressed in IPCs in vitro and in WB-A cells. Finally, Expression of amylase, PP, somatostatin and glucagon was not detected. Taken together, the changes in gene expression profiles of post-transplanted WB-A cells suggest a correlation of the sequence of gene activation similar to β-like cell differentiation and maturation. The degree of maturation and function of IPCs derived from transplanted WB-A cells in vivo might be more superior compared with induced IPCs in vitro.
Discussion
In this study, we report that WB cells can be induced efficiently into IPCs by a novel three-step protocol based on a combination of 5-AZA, TSA, ITS and RA and other maturation factors, after only 17 days in vitro. The research focused on inducing Pdx1-positive cells with an efficiency of over 5%, which was five times more than induction by the transgene. This contributed to the highly selective differentiation into functional β-like cells in vitro and in vivo.
Both 5-AZA and TSA were important for inducing WB cell dedifferentiation. 5-AZA, an inhibitor of DNA methylation, is important for differentiation processes, such as osteoblast23, cardiomyocyte13, and, more recently, regulation of the differentiation of pancreatic endocrine cells11. We sequentially treated the cells with the histone deacetylase inhibitor TSA based on the hypothesis that chromatin remodeling would enhance induction and further differentiation toward pancreatic lineage24. In our experiment, a combination of these factors might promote WB cell dedifferentiation, as confirmed by the specific morphological changes of spindle-shaped cells, which showed that dedifferentiation might have occurred in most cases25 and downregulation of the hepatocyte dedifferentiation marker C/EBPβ at the end of stage 1.
Another crucial step in our differentiation protocol was that RA was extremely important immediately after cell treatment with TSA for inducing the expression of Pdx1. Recently, RA was demonstrated to be important for the development of the early embryonic pancreas, specifically for promoting differentiation of Pdx1-expressing cells from mouse and human embryonic stem cells26,27,28. The unique feature of our induction strategy was using the combination of ITS and RA to activate specific pancreatic β-cell differentiation. We discovered that after ITS and RA induction, differentiated WB cells expressed pancreatic developmental markers, such as Pdx1, Ngn3 and NKX2.2, which are critical for the formation of pancreatic islet cells7. The reason why insulin release was not responsive to glucose stimulation may be due to the fact that the β-cell developmental genes Pax-4, Pax-6 and MafA and the functional gene GK were not detected in the WB-A cells.
Pdx1-expressing pancreatic precursor WB-A cells could be induced to differentiate into IPCs after the sequential gene expression of appropriate transcription factors, such as Ngn3, NeuroD, Nkx2.2, Pax4, Pax6 and MafA. Although Pdx1 could be the main genetic switch to transform hepatocytes into pancreatic endocrine cells, the exact mechanism remains to be elucidated. Furthermore, it is also unclear whether these small molecules transformed WB cells into a single type of β hormone-expressing cell. One possible reason could be that the Pdx1-expressing WB-A cells could be efficiently induced to differentiate into highly efficient pancreatic endocrine cells and eventually into IPCs in vitro or in vivo in a hyperglycemia microenvironment with regard to the stage of β cell differentiation. Not all WB-A cells terminally differentiate into mature phenotypes in vitro and some WB-A cells continue to be in the premature stage under these conditions. So we selected Pdx1-expressing WB-A cells in stage 2, rather than IPCs in stage 3, as better candidates for transplant cells to meet the induction needs in vivo.
Of note, the induced cells did not form clusters in the end stage and the pancreatic developmental gene NKX6.1 was not expressed in IPCs either in vivo or in vitro. While the pancreatic functional genes GK and Kir6.2 and the endocrine gene insulin II were upregulated in the end-induced cells in vivo compared with in vitro. Nkx6.1 has a highly β-cell restricted expression in adult rats29. Moreover, based on the gene-expression profile changes of the post-transplanted WB-A cells, we presume that some of the cells may still have remained in an immature state after induction. In addition, the induced IPCs derived in vivo might be more mature than those derived in vitro. It is possible to show that an in vivo hyperglycemia microenvironment more efficiently promotes WB-A cells to undergo differentiation into functional β-like IPCs, but this heterogeneity remains to be further investigated.
Influencing factors should be explored, including various combinations and concentrations of different small molecules and the timing and ways of application, along with animal models, immunologic rejection and the appropriate stage to detect cell expression after transplantation, long-term cellular engraftment and physiological insulin secretion. How long these induced cells survive in this differentiated state both in vitro and in vivo is worth exploring.
In summary, we have established an efficient approach to induce hepatic stem-like WB cells to differentiate into Pdx1-expressing pancreatic precursor cell using a chemical small molecule defined culture system. Some Pdx1-expressing cells could further differentiate into IPCs in vitro and in vivo. This newly established strategy could facilitate the future study of mechanisms and the discovery of drugs for hepatocyte-based therapies for diabetes in the field of pancreatic islet neogenesis and maturation.
Methods
Cell culture
WB cells17,30 (passage 16) were kindly provided by Dr. Liping Lv (The Academy of Military Medical Sciences, Beijing, China). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (PAA, Dresden, Germany), 11.1 mmol/L (mM) D-glucose, 100 U/ml penicillin and 100 μg/ml STZ (Sigma, Missouri, USA).
The rat insulinoma cell line INS-1 (passages 80), was from the People's Liberation Army General Hospital (PLA General Hospital) laboratory. These cells express and process rat insulin and are capable of insulin release in response to glucose stimulation. Cells were continuously maintained in our laboratory for one year in Roswell Park Memorial Institute (RPMI) 1640 medium (Sigma, Missouri, USA) and used as a positive control for studies of insulin content and insulin release.
Induction of WB cells into IPCs
We designed a differentiation protocol of three basic steps. In the first step (chromatin remodeling), WB cells were plated at a density of 2 × 105 per well of 6-well plates (Corning, New York, USA) in a basal medium containing knockout-serum DMEM and the main components: 1 mM β-mercaptoethanol, 1% non-essential amino acids, 1% B27 supplement, 2 mM L-glutamine, 2% N2 supplement (all from Invitrogen, California, USA), 20 ng/ml fibroblast growth factor (Peprotech, New Jersey, USA), 20 ng/ml epidermal growth factor (Peprotech, New Jersey, USA), 100 U/ml penicillin and 100 mg/ml STZ (Sigma, Missouri, USA). Cells were treated with 5 μmol/L (μM) 5-AZA (Sigma, Missouri, USA). After 48 hours, cells were treated with 100 nmol/L (nM) TSA (Sigma, Missouri, USA) for 24 hours. In the second step (induction and differentiation), basal medium was changed to induction medium containing DMEM with low glucose (1 g/L; Sigma, Missouri, USA), 1×ITS (Invitrogen, California, USA), 2 μM RA (Sigma, Missouri, USA) and the main components for 7 days. In the last step (maturation), the medium was modified from differentiation medium by adding 10 mM nicotinamide (Sigma, Missouri, USA) without fibroblast growth factor and epidermal growth factor for 7 days. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. For each stage, medium was changed every 2 days.
Semi-quantitative and quantitative RT-PCR
Total RNA was extracted by TRI Reagent (Fermentas, Burlington, ON, Canada) and reverse-transcribed using SuperScript III RT-PCR system (Invitrogen) according to the manufacturer's protocol16. 1 μl of cDNA sample was used for PCR amplification. Quantitative RT-PCR was performed using QuantiFast SYBR Green PCR Kit (Qiagen) and analyzed with DNA engine Opticon2 (MJ Research). The name and sequences of the semi-quantitative and the quantitative RT-PCR primers for each pair are listed in Supplementary table 1 and Supplementary table 2, respectively.
Glucose-stimulated insulin release assay in vitro
For each determination, cells were plated in 6-well plates at 2 × 105 per well. At stages 2 and 3, cells were incubated overnight in serum-free DMEM with low glucose (1 g/L). For insulin secretion assays, WB-A cells and IPCs were starved overnight in serum-free medium containing 0.5% bovine serum albumin (Serana, Bunbury Australian) and 5.5 mM glucose and stimulated with 25 mM glucose for 2 hours. The buffer was collected and frozen at −80°C until assay for insulin content. The amount of insulin released was determined using an ultrasensitive rat insulin enzyme-linked immunosorbent assay (ELISA) (Linco Research, Missouri, USA) according to the manufacturer's protocol.
TEM
Cell aggregates were fixed with 4% phosphate-buffered paraformaldehyde (Sigma, Missouri, USA) for 4 hours at 48°C and post-fixed in 1% osmium tetroxide (Sigma, Missouri, USA). Cells were harvested with a cell scraper after in vitro induction. Cell aggregates were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4, Sigma, Missouri, USA) for 1 hour at room temperature and post-fixed with 1% osmium tetroxide in 0.1 M cacodylate buffer (Sigma, Missouri, USA) for 1 hour at room temperature, dehydrated in ethanol and embedded in Epon 812 (Aurion, Wageningen, the Netherlands). After polymerization, the plastic was removed and ultrathin sections were cut parallel and perpendicular to the flask surface. Thin sections were counterstained with uranyl acetate and lead nitrate and examined with a HitachiH-7650 transmission electron microscope (Hitachi, Olympus Japan).
Cell transplantation and nephrectomy
The Institutional Animal Care and Use Committee of the PLA General Hospital approved all animal procedures. The 8- to 10-week-old nude rats (PLA General Hospital Animal Centre) were made hyperglycemic by a single intraperitoneal injection of STZ (Sigma, USA) at 65 mg/kg body weight31. When blood glucose reached levels > 16.7 mmol/L, 1 × 106 WB-A cells/rat (n = 6) were transplanted into the left renal subcapsular space. Control rats receiving 1 × 106 WB (n = 6) cells/rat in 50 μl PBS and negative controls received 50 μl PBS (n = 3). Fasting blood glucose levels were measured every 3 days in samples obtained from the tail vein using a blood glucose test meter and strips (One Touch Ultra Vue, USA). Three of the six WB-A cell-implanted rats underwent nephrectomy at day 36. The others underwent nephrectomy at day 60 posttransplantation to assess metabolic activity of the transplanted WB-A cells. The explanted tissues containing implanted WB-A cells were analyzed for gene expression and morphology evaluation. Most of the diabetic rats that underwent sham surgery and had transplanted WB cells without insulin treatment died approximately at day 32 to 39 posttransplantation.
IPGTT and glucose-stimulated insulin release assay in vivo
At day 60 posttransplantation nude rats with WB-A cells (n = 3), DM rats (n = 3) and normal nondiabetic rats (n = 5 control) were fasted for 6 h by transfer to a clean cages without food at the end of the dark (feeding) cycle. Nude rats were weighed. After 6 h of fasting, Fasting glucose and insulin levels were obtained from venous blood from small tail clips. Then, three groups injected 2 g/kg body glucose (Sigma, USA) intraperitoneally. Blood glucose values were obtained at 15, 30, 60, 90, 120 min. Insulin values were obtained at 30, 60, 90, 120, 150, 180 min.
Histology and immunofluorescence
Explanted tissues and rat pancreas tissues were fixed by immersion in 4% paraformaldehyde for 48 hours at 4°C. Samples were placed overnight in a 30% sucrose solution and embedded with embedding medium. Sections were stained with HE. Cytospin slides were made from cultured WB-A cells, pancreas tissues with 4% paraformaldehyde for 15 min and blocked with 5% bovine serum albumin and 0.5% Triton X-100 in PBS. Cells and sections were incubated with primary antibodies: guinea pig anti-rat insulin (1:100) (Sigma, Missouri, USA), goat anti-rat Pdx1 (1:100) (Santa Cruz, California, USA) antibody overnight at 4°C, followed by Alexor-594-conjugated goat anti-guinea pig polyclonal antibody (1:500) (Sigma, USA) and tetramethylrhodamine isothiocyanate-conjugated donkey anti-goat polyclonal antibody (1:200) (Santa Cruz, California ,USA) at room temperature in the dark for 2 hours. Nuclei were counterstained with Hoechst-33342 reagent (1:3000) (Sigma, California, USA). Images were captured using an Olympus phase contrast fluorescent microscope (Olympus IX-71, Olympus, Japan).
Statistical analyses
Data are presented as means ± s.e.m. Statistical differences were measured using an independent sample t-test. Statistical significance was set at probability (P) < 0.05 or P < 0.01.
References
Shapiro, A. M. et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 343, 230–8 (2000).
Ameri, J. et al. FGF2 specifies hESC-derived definitive endoderm into foregut/midgut cell lineages in a concentration-dependent manner. Stem Cells 28, 45–56 (2010).
Lammert, E., Cleaver, O. & Melton, D. Role of endothelial cells in early pancreas and liver development. Mech Dev 120, 59–64 (2003).
Nakajima-Nagata, N. et al. In vitro induction of adult hepatic progenitor cells into insulin-producing cells. Biochem Biophys Res Commun 318, 625–30 (2004).
Ber, I. et al. Functional, persistent and extended liver to pancreas transdifferentiation. J Biol Chem 278, 31950–7 (2003).
Park, I. H. et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451, 141–6 (2008).
Kawasaki, H. et al. In vitro transformation of adult rat hepatic progenitor cells into pancreatic endocrine hormone-producing cells. J Hepatobiliary Pancreat Surg 15, 310–7 (2008).
Takahashi, K. Cellular reprogramming-lowering gravity on Waddington's epigenetic landscape. J Cell Sci (2012).
Nakagawa, M. et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26, 101–6 (2008).
Wang, H. S. et al. Transplantation of insulin-producing cells derived from umbilical cord stromal mesenchymal stem cells to treat NOD mice. Cell Transplant 20, 455–66 (2011).
Lefebvre, B. et al. 5′-AZA induces Ngn3 expression and endocrine differentiation in the PANC-1 human ductal cell line. Biochem Biophys Res Commun 391, 305–9 (2010).
Chang, S. et al. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 24, 8467–76 (2004).
Tomita, S. et al. Autologous transplantation of bone marrow cells improves damaged heart function. Circulation 100, II247–56 (1999).
Cai, J. et al. Generation of homogeneous PDX1(+) pancreatic progenitors from human ES cell-derived endoderm cells. J Mol Cell Biol 2, 50–60 (2010).
Ostrom, M. et al. Retinoic acid promotes the generation of pancreatic endocrine progenitor cells and their further differentiation into beta-cells. PLoS One 3, e2841 (2008).
Yang, L. et al. In vitro trans-differentiation of adult hepatic stem cells into pancreatic endocrine hormone-producing cells. Proc Natl Acad Sci U S A 99, 8078–83 (2002).
Chen, S. et al. A small molecule that directs differentiation of human ESCs into the pancreatic lineage. Nat Chem Biol 5, 258–65 (2009).
Johannesson, M. et al. FGF4 and retinoic acid direct differentiation of hESCs into PDX1-expressing foregut endoderm in a time- and concentration-dependent manner. PLoS One 4, e4794 (2009).
Shen, C. N., Slack, J. M. & Tosh, D. Molecular basis of transdifferentiation of pancreas to liver. Nat Cell Biol 2, 879–87 (2000).
Shafritz, D. A., Oertel, M., Menthena, A., Nierhoff, D. & Dabeva, M. D. Liver stem cells and prospects for liver reconstitution by transplanted cells. Hepatology 43, S89–98 (2006).
Tateno, C. & Yoshizato, K. Long-term cultivation of adult rat hepatocytes that undergo multiple cell divisions and express normal parenchymal phenotypes. Am J Pathol 148, 383–92 (1996).
Yamada, S. et al. In vitro transdifferentiation of mature hepatocytes into insulin-producing cells. Endocr J 53, 789–95 (2006).
Vaes, B. L. et al. Inhibition of methylation decreases osteoblast differentiation via a non-DNA-dependent methylation mechanism. Bone 46, 514–23 (2010).
Haumaitre, C., Lenoir, O. & Scharfmann, R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol Cell Biol 28, 6373–83 (2008).
Chen, Y., Wong, P. P., Sjeklocha, L., Steer, C. J. & Sahin, M. B. Mature hepatocytes exhibit unexpected plasticity by direct dedifferentiation into liver progenitor cells in culture. Hepatology 55, 563–74 (2012).
Micallef, S. J. et al. Retinoic acid induces Pdx1-positive endoderm in differentiating mouse embryonic stem cells. Diabetes 54, 301–5 (2005).
Shi, Y. et al. Inducing embryonic stem cells to differentiate into pancreatic beta cells by a novel three-step approach with activin A and all-trans retinoic acid. Stem Cells 23, 656–62 (2005).
D'Amour, K. A. et al. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nat Biotechnol 24, 1392–401 (2006).
Delisle, J. C. et al. Pdx-1 or Pdx-1-VP16 protein transduction induces beta-cell gene expression in liver-stem WB cells. BMC Res Notes 2, 3 (2009).
Fujikawa, T. et al. Teratoma formation leads to failure of treatment for type I diabetes using embryonic stem cell-derived insulin-producing cells. Am J Pathol 166, 1781–91 (2005).
Sulochana, K. N., Punitham, R. & Ramakrishnan, S. Beneficial effect of lysine and amino acids on cataractogenesis in experimental diabetes through possible antiglycation of lens proteins. Exp Eye Res 67, 597–601 (1998).
Acknowledgements
This research was supported in part by National Basic Science and Development Programme (2012CB518103) and the 863 Projects of Ministry of Science and Technology of China (2011AA020113) and National Natural Science Foundation of China (No. 81230061 and 31270820).
Author information
Authors and Affiliations
Contributions
J. Liu designed and conducted the experiments, performed data analyses and wrote the manuscript; H. Hao designed and conducted the experiments; and Y. Liu carried out the experiments and analyzed data; H. Wang conducted RT-PCR experiments and analyzed data; Q. Han designed and conducted the experiments; Y. Mu contributed to review the manuscript; W. Han contributed to design and conduct the experiments, reviewed and revised the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Liu, J., Liu, Y., Wang, H. et al. Direct differentiation of hepatic stem-like WB cells into insulin-producing cells using small molecules. Sci Rep 3, 1185 (2013). https://doi.org/10.1038/srep01185
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep01185
This article is cited by
-
Combination of the modulators of epigenetic machinery and specific cell signaling pathways as a promising approach for cell reprogramming
Molecular and Cellular Biochemistry (2022)
-
Small-molecule-mediated reprogramming: a silver lining for regenerative medicine
Experimental & Molecular Medicine (2020)
-
Current progress in hepatic tissue regeneration by tissue engineering
Journal of Translational Medicine (2019)
-
Cellular reprogramming for pancreatic β‐cell regeneration: clinical potential of small molecule control
Clinical and Translational Medicine (2014)
-
Research status and prospect of stem cells in the treatment of diabetes mellitus
Science China Life Sciences (2013)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
