
Abstract
Chronic treatment with glucocorticoids increases the mass of adipose tissue and promotes metabolic syndrome. However little is known about the molecular effects of dexamethasone on adipose biology. Here, we demonstrated that dexamethasone induces progenitor cells to undergo adipogenesis. In the adipogenic pathway, at least two cell types are found: cells with the susceptibility to undergo staurosporine-induced adipose conversion and cells that require both staurosporine and dexamethasone to undergo adipogenesis. Dexamethasone increased and accelerated the expression of main adipogenic genes such as pparg2, cebpa and srebf1c. Also, dexamethasone altered the phosphorylation pattern of C/EBPβ, which is an important transcription factor during adipogenesis. Dexamethasone also had effect on mature adipocytes mature adipocytes causing the downregulation of some lipogenic genes, promoted a lipolysis state and decreased the uptake of glucose. These paradoxical effects appear to explain the complexity of the action of glucocorticoids, which involves the hyperplasia of adipose cells and insulin resistance.
Similar content being viewed by others

Introduction
Metabolic imbalance is connected to hypertrophic and/or hyperplastic obesity. Hyperplastic obesity is due to an enhanced recruitment and commitment of progenitor cells toward a terminally differentiated adipocyte phenotype. A better understanding of the early molecular processes involved in adipogenesis is a basic concept in adipose biology that has importance in clinical settings.
The study of differentiation processes has been highly facilitated by cell culture models that use specific molecules to promote the various differentiation programs. 3T3-F442A cells, which are derived from murine embryonic 3T3 cells, develop morphological characteristics of mature white adipocytes both in vivo and in vitro; these characteristics include a spherical shape, an accumulation of triglycerides and the expression of adipocyte-specific markers1,2,3,4. The 3T3-F442A cell line is a clone that was isolated from the 3T3 Clone18 line to ensure single-cell clone characteristics5. The 3T3-F442A clone differentiates in vitro into adipocytes and injection of these preadipocytes into nude mice give rise to fat pads that exhibit histological features of adipose tissue, indistinguishable from the host's normal adipose tissue, demonstrating that they respond to in vivo physiological signals2,4. Similar pads were not observed when nude mice were injected with the sister 3T3-L1 cell line4. These results make the 3T3-F442A cells a suitable model for studying adipose differentiation and molecules that could regulate it.
To undergo adipose differentiation, confluent 3T3-F442A cells require approximately 48 h in a medium supplemented with fetal bovine serum (FBS), which typically has high adipogenic activity6, whereas 3T3-L1 cells additionally require methyl isobutyl xanthine (MIX) and dexamethasone (Dex) in FBS medium7,8. If 3T3-F442A cells are cultured in the absence of fetal bovine serum or growth hormone (non-adipogenic media), the cells do not undergo adipose differentiation6.
Because 3T3-F442A cells respond to physiological differentiation signals in the body2, these cells are an adequate model for the study of the effect of glucocorticoids on adipocyte cell biology and metabolism. We have previously reported that in serum-free media or the absence of additional adipogenic factors, low concentrations of staurosporine (St), which is a serine/threonine kinase inhibitor, rapidly induce 3T3-F442A cells to undergo adipogenesis through the induction of GSK3β activity9. We also demonstrated that early adipogenesis includes two identifiable stages; the first stage involves the induction of adipogenesis by St (0–4 h) and the second stage, which occurs in the absence of St, is the stabilization stage (4–48 h), at the end of which (44 h after the initiation of St induction), the cell is stable. This 44-h timeframe is susceptible to manipulation by various substances that block adipogenesis, which would return the cells to the initial state such that the cells would need to be re-induced to undergo differentiation9. After these two stages, the cells enter clonal expansion and express the adipose phenotype10. The identification of the two stages of early adipogenesis makes it possible to study the early molecular events that regulate both the induction and stabilization stages of the adipogenic process in more detail, which includes the identification of the participating genes and the analysis of the effect of different drugs on these processes.
Other studies have shown that high glucocorticoid levels can cause metabolic syndrome in animal models11. As shown in a genome-wide analysis, these compounds appear to modify the gene expression network that is involved in triglyceride homeostasis12. However, the changes that occur in response to glucocorticoid treatment are poorly understood. Chronic treatment with glucocorticoids, such as Dex, induces obesity and metabolic syndrome, which impairs the adipose tissue metabolism such that it resembles the metabolism observed in individuals with Cushing's syndrome13,14. These effects may persist after treatment with glucocorticoid is terminated, which demonstrates its severity15. Adipose tissue comprises several cell types, including preadipocytes and terminally differentiated (mature) adipocytes. Thus, the in vivo administration of Dex in an animal model makes it difficult to differentially study the action of glucocorticoids in preadipocytes and adipocytes because many of the biological effects are combined in the adipose tissue; in addition, other organs and tissues are involved. A cell culture system allows for separate study of the action of substances in preadipocytes and mature adipocytes. Thus, it is possible to define the biological effects that are exerted by glucocorticoids during early adipogenesis and in mature adipocyte lipid metabolism.
In this study, we utilize 3T3-F442A cells in culture, the defined stages of adipogenesis and the formation of terminally differentiated adipocytes to study the action of Dex on the early stages of adipose differentiation and the lipid metabolism of mature adipocytes. We analyzed the expression of pparg2 and cebpa, which are two well-known genes in adipogenesis16,17,18, the induction of the early adipogenesis transcription factors cebpb and srebf1a10,19 and the induction of srebf1c, which is involved in the regulation of the expression of lipogenic genes20. The transcription factor C/EBPβ participates in the transactivation of pparg2 and cebpa21 and appears to be related to the clonal expansion of these cells22,23. In 3T3-L1 cells induced with adipogenic serum and MIX/Dex, cebpb is expressed early during the adipogenic program19. However, the acquisition of the DNA binding activity of cebpb and transactivation of pparg2 and cebpa are delayed more than 14 h24. We recently discovered that cebpb is induced early and is transiently expressed and that its protein (C/EBPβ) is phosphorylated at the Thr188 residue through the activity of GSK3β, which ultimately leads to the transactivation of srebf1a10.
Our results indicate that Dex enhances the recruitment of 3T3-F442A progenitor cells toward adipogenesis. In mature adipocytes, Dex decreased both the lipogenic gene expression and glucose uptake and increased lipolysis. Our results show that Dex might have a complex dual role in the impairment of adipose tissue homeostasis: 1) it stimulates the differentiation of preadipocytes into adipocytes; and, 2) alters the lipid metabolism and insulin sensitivity of differentiated fat cells. These actions might lead to obesity and impaired lipid homeostasis and induce insulin resistance in an organism, which are 3 of the main symptoms of metabolic syndrome.
Results
Dex increases the recruitment of adipose cell progenitors in a cell culture of 3T3-F442A cells
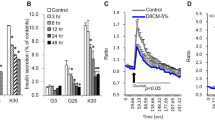
To determine the effect of Dex on the adipogenesis of 3T3-F442A cells, post-confluence cultures were incubated with non-adipogenic media with different concentrations of Dex and 10 nM St for 4 h. After 4 h, the cultures were transferred to non-adipogenic media and incubated for an additional 140 h to allow the cells to reach terminal differentiation and form mature adipocytes. To quantify the extent of adipogenesis, we counted the number of adipose clusters at the end of the experiments. The cells that were induced to differentiate upon treatment formed adipose clusters; thus, the number of adipose clusters corresponds with the number of induced progenitor cells. The administration of 0.25 μM Dex increased the number of adipose clusters by 2-fold when the glucocorticoid was added either simultaneously with St during the induction stage (first 4 h) or stabilization stage (4–48 h, after the removal of St. Figure 1A, B). Dex also promoted the formation of larger clusters of adipocytes, which suggests that it also enhanced the selective amplification of the induced cells (Figure 1C). The addition of Dex before treatment with St did not increase the number of adipose clusters, even if the cells were allowed to incubate in the presence of Dex for up to 8 h (Figure 1C). The administration of Dex alone, even at concentrations as high as 1 μM, did not promote adipose conversion (Figure 1A). These results demonstrate that Dex does not induce adipogenesis but only enhances its induction by St, which suggests that Dex might increase the recruitment of progenitor cells toward the differentiation process. We performed experiments to determine the colony formation ability and differentiation. We seeded 3T3-F442A cells at a low density (200 cells per 100-mm dish) in non-adipogenic media until the cells formed large colonies that were confluent at the center of the colony but did not undergo adipose conversion. We then induced adipogenesis through the addition of St in the absence or presence of 0.25 μM Dex for 4 h. The cultures were then transferred to non-adipogenic media and monitored for an additional 192 h, at which point the cells were fixed and stained with Oil Red O. The results show that the total colony formation ability of 3T3-F442A cells was approximately 50% in all culture conditions (Figure 1D). The cultures under non-adipogenic conditions exhibited only a low number of adipose colonies (18% of the total colonies), whereas the cultures to which only St was added formed 31% of the total adipose colonies (Figure 1D). The cultures induced with St for 4 h and treated with Dex during either the induction stage or stabilization stage formed approximately 53% of the adipose colonies; this amount is similar to the percentage observed in cultures incubated in the highly adipogenic FBS media (Figure 1D), which promotes maximum adipose conversion of 3T3-F442A cells6. The incubation of the cells with Dex alone did not increase the number of adipose colonies (Figure 1D). When we quantified the number of adipose clusters per adipose colony, we found that more than 90% of the adipogenic colonies were formed by a single adipose cluster at the center of the colony (Figure 1E). This finding further supports the conclusion that each adipose cluster arises from the selective multiplication of a single adipose parental cell. These results suggest that Dex can recruit progenitor cells toward adipose differentiation only if St induces the cells during or before treatment with the glucocorticoid. Because constant incubation of 3T3-F442A cells with FBS promoted the same proportion of adipose conversion as the St/Dex treatment (Figure 1F), we concluded that the treatment with the combination of St and Dex for 4 h during the induction stage is sufficient to recruit the same proportion of susceptible progenitor cells toward adipogenesis as obtained with FBS treatment. These results also demonstrate that 3T3-F442A cells comprise at least two progenitor or preadipocyte cell types that are able to undergo adipogenesis; one group of cells can be induced by St alone and the other group is recruited by both Dex and St. Therefore, Dex can recruit additional progenitor cells toward adipogenesis and enhances the clonal amplification of the induced cells, which suggests that these two mechanisms would exert a hyperplastic state of adiposity.

Dex increases the number of adipose clusters in 3T3-F442A cells induced to undergo adipogenesis by St.
(A). Effect of the Dex concentration on adipose differentiation. Post-confluence 3T3-F442A cells were incubated with St and various concentrations of Dex for 4 h. Parallel cultures were either incubated with 1 μM Dex for 4 h or maintained in non-adipogenic medium. (B). Induction of adipogenesis by St in the presence or absence of Dex. (C). Adipose cluster formation. Representative photographs of 3T3-F442A cell cultures treated with St and Dex are shown. The images show the adipose cell clusters. (D). Recruitment of adipose progenitors. The analysis was performed using a colony-based assay. (E). Representative photograph of the colony-based assay used to evaluate the effect of Dex on the recruitment of new adipose progenitors. (F). Comparison of the adipogenesis induced by the St/Dex mixture with that induced by a highly adipogenic serum. (G). Effect of RU486 on the recruitment of adipogenic progenitors. All of the results are representative of 3 independent experiments. The asterisks in the plots indicate significant differences (with a P ≤ 0.05). N-Ad, non-adipogenic treatment; St, staurosporine; Dex, dexamethasone; FBS, fetal bovine serum.
We also tested the effect of RU486, which is a well-known antagonist of Dex that has been reported to block the action of Glucocorticoid Receptor (GR)25. A similar number of adipose clusters were found in the cultures treated with St/Dex in the absence or presence of RU486 (Figure 1G). Importantly, the cultures treated with only St and RU486 (in the absence of Dex) also underwent an adipose differentiation that was similar to that observed in the cultures that were treated with St/Dex (Figure 1G), suggesting that this compound also enhanced adipose differentiation similarly to Dex. It is interesting to note that the D1 cell line, which differentiates into osteoblasts and adipocytes, also underwent adipose conversion in the presence of this glucocorticoid antagonist26. These results are not surprising since the activation or repression of specific genes occurs through the GR and that selective ligands would lead to positive or negative effects (Please see Discussion).
Our results raise the possibility that the 3T3-F442A cell line comprises a group of cells with certain characteristics of adipose progenitors and stem cells-like that can be induced to differentiate by defined small molecules. One of the characteristics of a stem cell is its potential to renew itself in each division cycle, thereby maintaining a similar proportion of stem cells during serial cultivation and terminal differentiation. If this hypothesis were correct, it would be expected that cultures that are terminally differentiated into adipocytes should maintain a certain proportion of adipose cell precursors. To test this hypothesis, we studied cultures of terminally differentiated adipocytes. We seeded the cells from these terminally differentiated adipose cultures into indicator dishes at cloning cell densities and treated them with adipogenic conditions as described above. We performed these experiments with 5 subsequent culture transfers. Throughout these culture transfers, the cells were capable of continuous subculture and gave rise to a constant proportion of differentiated colonies depending on the adipogenic conditions used (Figure 2A). Because the number of differentiation-susceptible cells remained constant throughout the subcultures and no depletion in their cloning efficiency and adipogenic potential was observed (Figure 2A), we determined that the 3T3-F442A clones comprise a population of cells with the ability to give rise to two daughter cells; one daughter cell is committed to the adipose lineage and the other has the capability of self-renewal, which means that it remains a progenitor or stem cell-like even in highly differentiation-inducing culture conditions. The 3T3 cell cultivation protocol used by others and by us in previous adipogenesis studies involves the culture of 3T3 cell stocks and the transfer of these stocks prior to their confluence to avoid differentiation. These conditions would not reveal the possible stem-like characteristic of this 3T3 cell line.

Stem cell-like characteristics of 3T3-F442A clone.
(A). Cells from the terminally differentiated adipose cultures were detached with trypsin and the lipid-containing cells were evaluated by dark-field microscopy to determine the percentage of adipose cells. The cells were seeded into indicator dishes at cloning cell densities of 100 cells per 100-mm tissue culture dish. The cultures were then incubated under adipogenic conditions as described in Materials and Methods. We performed these experiments with 5 subsequent culture transfers. At the end of the cultures transfers, we determined the colony formation ability and proportion of adipose colonies. (B). We performed clone isolation experiments from the parental 3T3-F442A cell line. We seeded cells at a density of approximately 500 cells per 100-mm tissue culture dish and allowed the cells to grow for 10 days in non-adipogenic media. When the centers of the colonies reached confluence but were still well separated from each other, we treated the cells with St for 4 h and then changed media to non-adipogenic media. When the colonies began to show the first sign of adipose conversion, we selected 3 colonies that showed adipogenic susceptibility and individually trypsinized and expanded these into independent new indicator cultures. We seeded these cultures at normal densities and determined the percentage of adipose conversion induced by St in the presence or absence of Dex.
We also performed clone isolation experiments using the parental 3T3-F442A cell line. We seeded the cells at a cell density of approximately 500 cells per 100-mm tissue culture dish and allowed them to grow for 10 days in non-adipogenic media. When the centers of the colonies reached confluence but were still well separated from each other, we treated the cells with St for 4 h and then transferred them to non-adipogenic media. When the colonies began to show the first sign of adipose conversion, we selected 3 colonies that showed adipogenic susceptibility and individually trypsinized and expanded these in separate new indicator cultures. We seeded these cultures at normal densities and tested the adipose conversion induced by St in the absence or presence of Dex. The 3 clones (3T3-F442A-St1, -St2 and -St3) underwent adipose conversion similarly to the 3T3-F442A parental line (Figure 2B). In all cases, Dex increased the adipose conversion to a similar extent as in the parental clone (3T3-F442A). This finding demonstrates that these clones had the same susceptibility to Dex-induced progenitor recruitment (Figure 2B), which suggests that this characteristic is clonally preserved and inherited. These results indicate a possible stem-like characteristic in subpopulations of the 3T3-F442A clone and warrants further investigation to explore the presence of cells with specific stem cell markers through cell sorting experiments.
Glucocorticoid differentially regulates the adipogenic gene expression during the early stages of adipogenesis
We evaluated the adipogenic gene expression by relative RT-PCR in post-confluence cells that were induced to differentiate by St in the presence or absence of Dex. The expression level of pparg2 decreased during the first 4 h of St/Dex treatment (induction stage); this decrease was followed by a significant increase that was evident within 8 h of the initiation of the St/Dex incubation (Figure 3A, pparg2 inset). However, pparg2 expression in cultures treated only with St remained constant (at its basal level) for the first 30 h, after which expression began to increase significantly (Figure 3A).

Dex alters the expression profiles of adipogenic genes and phosphorylation of C/EBPβ during adipogenesis.
(A). Post-confluence cultures were induced with St or St/Dex for 4 h or with St for 4 h and then with Dex for the following 26 h. The total RNA was extracted and the expression levels of pparg2, cebpa, cebpb, srebf1a and srebf1c were determined by RT-PCR. The data show the mean fold change (compared with the baseline level at time = 0) ± S.D. of 3 independent experiments. (B). Immunoblotting of the phosphorylation of the Thr188 residue of the C/EBPβ transcription factor. The immunoblots were challenged with anti-phospho C/EBPβ Thr188 and anti-C/EBPβ antibodies; an anti-GDI antibody was used as the loading control.
In the cultures that were induced with St in the absence or presence of Dex, cebpa expression remained constant for the first 30 h and then increased more significantly with Dex treatment (Figure 3A). However, Dex did not induce the early increase in cebpa expression that was observed in the analysis of pparg2 expression during adipogenesis.
The expression levels of srebf1a increased similarly during the first 4 h in the presence and absence of Dex. However, within 8 h, a faster and larger increase in the expression of this gene was observed in the cultures treated with Dex; this increase was faster than that observed for srebf1c (Figure 3A). In all cases, all of the studied genes reached their maximum expression levels in the terminally differentiated cultures within 144 h of cultivation (Figure 3A).
We found that the expression levels of cebpb were similarly increased in cultures treated with St in the presence or absence of Dex (Figure 3A), which indicates that the expression of this gene is not modulated by Dex. In the adipogenic program, the phosphorylation of the Thr188 residue of C/EBPβ is important and our results showed that Dex increased the phosphorylation of this transcription factor (Figure 3B). These findings demonstrate that in addition to the recruitment of progenitor cells toward adipogenesis, Dex promotes a rapid increase in pparg2 transcription and modulates the phosphorylation of C/EBPβ during early adipogenesis. The expression of the other adipogenic genes did not show an early increase in the presence of Dex but did reach a higher level in the cultures treated with Dex.
Glucocorticoid impairs lipid homeostasis and glucose uptake in mature adipocytes
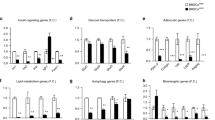
The effects of glucocorticoids have been extensively studied in animal models, which show that these molecules lead to lipid accumulation in liver cells27,28 and have a lipogenic effect on adipose tissue29. Glucocorticoids also induce weight gain and obesity and impair glucose tolerance30,31. Because these in vivo effects of glucocorticoid in animal models are the result of a combination of many biological actions in several organs and tissues and because adipose tissue is composed of various cell types, it is difficult to characterize the action of glucocorticoids in mature adipocytes. Because adipocytes have a central role in mammalian lipid homeostasis, we studied the effect of Dex on the expression of adipogenic genes in terminally differentiated adipocyte cultures. We incubated cultures for 4 h with St to induce adipogenesis and then incubated the cultures in non-adipogenic media for an additional 140 h until the cells achieved terminal adipocyte differentiation. Over 90% of the cells in these cultures were adipocytes. After the cells reached terminal differentiation, the cultures were supplemented with or without 0.25 μM Dex for an additional 24 h. The results showed that the addition of this glucocorticoid to the adipocytes rapidly and significantly reduced the expression level of pparg2, cebpa, srebf1c and fabp4 but not that of srebf1a (Figure 3A), which demonstrates that Dex downregulates lipogenesis. In terminally differentiated cells that were induced to differentiate by FBS or the St/Dex mixture, these genes showed the same changes in expression levels as the cultures that were treated with Dex (data not shown).
Leptin and adiponectin are well-established hormones of adipocytes. In vivo studies have shown that leptin synthesis and secretion are increased in patients with Cushing's syndrome32, whereas the synthesis of adiponectin is decreased in these individuals compared with the level observed in non-obese subjects33. These changes have been associated with insulin resistance and metabolic syndrome34. In 3T3-F442A adipocytes treated with Dex, the expression levels of both the leptin (lep) and adiponectin (adipoq) genes decreased rapidly to low levels (Figure 4A), which reflect the downregulation of these genes. These data show that Dex does not increase lep expression in mature adipocytes, as was expected from the in vivo studies, but decreased its expression similarly to that of the other adipogenic genes. Thus, the increase in leptin synthesis and secretion that is observed in subjects treated with Dex appear to be contradictory; therefore, some other explanation should be offered to account for the discrepancy between our results and those obtained with in vivo systems. However, our data does show a decrease in the expression levels of the adipogenic transcription factors that regulate lep and adipoq synthesis, such as PPARγ2 and C/EBPα. Both of these factors increase the expression of adipoq and lep during adipogenesis. In mature adipocytes, Dex decreased the expression levels of pparg2 and cebpa, as shown above. RU486, the glucocorticoid receptor antagonist, also inhibited the adipogenic gene expression that is induced by Dex in mature adipocytes (Figure 4A).

Dex impairs lipid homeostasis in mature 3T3-F442A adipocytes.
Adipose conversion in post-confluence 3T3-F442A cells was induced by St for 4 h. The cells were then transferred to non-adipogenic media for up to 144 h until the cells attained terminal differentiation. Then, cultures were treated with 0.25 μM Dex. (A). Expression of adipose genes in mature adipocytes. Mature adipocytes were treated for 24 h with Dex and data represent the mean fold change (compared to the gene expression in the absence of Dex) ± S.D. of 3 independent experiments (n = 9). All of the data were obtained at 168 h (24 h after Dex) except the adipoq expression data, which were obtained at 216 h (72 h after Dex). (B). Induction of lipolysis in mature adipocytes by Dex. Cultures of mature adipocytes were treated with Dex or the GR inhibitor RU486 or a mix Dex + RU486 and 24 h later free glycerol was quantitated. Cultures treated with db-cAMP were used as a lipolysis positive control. Results are presented as a mean ± S.D. of 2 independent experiments (n = 6). (C). Effect of Dex on the adipocyte uptake of NBDG. The 3T3-F442A adipocytes were cultured for 5 days in adipogenic medium with or without 0.25 μM Dex. The cells were incubated for 60 min with 80 μM NBDG and 100 mM insulin. After the 60-min incubation, the free NBDG was removed from the cultures and the fluorescence associated with the cell monolayers was measured. The results are presented as the mean ± S.D. of 2 independent experiments (n = 16). (D). Effect of Dex on the number of adipose clusters after 3T3-F442A cell differentiation. Mature adipocytes were treated with Dex, then, cultures were followed for up to 96 h after Dex treatment and adipose clusters were counted. Data is presented as mean ± S.D. of 3 independent experiments (n = 9).
Because Dex reduced lipogenesis, we decided to study whether this glucocorticoid would also alter lipolysis, which is the other main pathway for the regulation of lipid metabolism. The adipocyte cultures were treated for 24 h with Dex or dB-cAMP, which is a well-known lipolytic agent used as a positive control. Lipolysis was determined as the amount of glycerol released into the cell culture media. The results showed that Dex significantly stimulated lipolysis but to a lesser extent than dB-cAMP (Figure 4B). RU486, the glucocorticoid receptor antagonist, also inhibited the lipolytic action of Dex (Figure 4B). These results demonstrate that Dex not only decreases lipogenesis but also stimulates adipocytes toward a lipolytic state through the GR.
The decreased lipogenic gene expression and increased lipolysis suggests that Dex could also induce a state of insulin resistance in adipocytes. Although previous reports have shown that chronic exposure to glucocorticoid results in insulin resistance14, it has not been clarified whether these compounds directly affect the effect of insulin in fat cells. We thus performed experiments in which we determined the amount of glucose transport through the uptake of fluorescent 2-deoxy-glucose (NBDG) by mature adipocytes. The addition of Dex to parallel cultures as described above showed that the uptake of NBDG decreased in approximately 40% of the insulin-treated adipocytes (Figure 4C), which shows that Dex induced insulin resistance in these cells. It is important to note that the Dex treatment of the adipocyte cultures for up to additional 96 h did not decrease the number of adipose clusters (Figure 4D).
Discussion
Obesity is a major health problem because of the excess body weight and its associated diseases, such as metabolic syndrome, which comprises insulin resistance, type-2 diabetes mellitus, cardiovascular disease and cancer. Recent data have suggested that changes in adipocyte differentiation and metabolism may initiate these risk factors.
The 3T3-F442A cell line offers an advantageous model to study adipogenesis and its regulation, since these cells respond to physiological signals of the body2,4. Our results offer several findings on the effect and mechanism of glucocorticoids on adipose biology. First, the 3T3-F442A cell line is composed of preadipocytes and progenitor cells that can be recruited to differentiate into adipocytes. In addition, although Dex alone does not induce adipogenesis, Dex can induce the recruitment of a greater number of adipose progenitors toward adipogenesis. Second, 3T3-F442A cells appear to comprise a subpopulation of cells with some stem cell-like characteristics. The presence of this population would explain the arising of two possible types of adipogenic progenitors. The 3T3-F442A cells are not only able to undergo adipogenesis but also differentiate into osteoblasts when exposed to other inducers, such as BMP2 and retinoic acid35, which is a blocker of adipose conversion during the stabilization stage9. Therefore, the putative stem cells of this cell clone might have the capability to respond to different inducers and commitment pathways. Third, Dex modulates the adipogenic gene pparg2 by increasing its expression early during adipogenesis but does not affect the expression of srebf1a, cebpb, cebpa and srebf1c. However, Dex stimulates C/EBPβ Thr188 phosphorylation, which is considered a key protein modification for adipogenesis10,36. Fourth, the expression levels of these adipogenic genes, with the exception of cebpb, in terminally differentiated cultures treated with Dex are higher than the levels obtained in cultures not treated with Dex during the early stages of adipogenesis. This result is not surprising because cultures treated with Dex have a higher number of adipose clusters. Fifth, Dex decreased the expression levels of the lipogenic genes pparg2, srebp1c, cebpa and fabp4 in mature adipocytes, which likely leads to reduced lipogenesis. Sixth, Dex decreased the expression levels of the adipokines lep and adipoq, which are 2 adipocyte hormones that exert systemic signaling effects. Seventh, Dex promoted a lipolytic state and insulin resistance in adipocytes. These results from cultures of 3T3-F442A cells allow to hypothesize that glucocorticoids have myriad paradoxical complex effects on adipose tissue that likely contribute to the development of metabolic syndrome. The action of Dex on the recruitment of adipose progenitor cells and the increase of lipolysis and reduction of lipogenesis in mature adipocytes appears to occur through selective mechanisms that involve the GR. It was recently shown that the activation or repression of specific genes occurs through the GR and that selective ligands can induce a subset of GR activity. These selective ligands act as chemical modulators at the level of GR-regulated promoters, which would lead to beneficial and harmful effects, depending in part on tissue-specific factors and the signaling pathways that are initiated37.
At least two types of cells are found in the 3T3-F442A cell line. The first group of cells can be induced to undergo adipose conversion by St alone (in the absence of Dex), whereas the second group requires both St and a glucocorticoid for the induction of adipogenesis (Figure 5A). We can suggest that, because two types of adipogenic cells were found in this clone, the 3T3-F442A cells might be used as a model cell culture that includes more than one adipose phenotype that could allow the analysis of adipose metabolic diversity. For example, in a culture of C3H10T1/2 cells, myostatin induced the formation of small and apparently immature adipocytes with metabolic activity that differs from that induced by a mixture of insulin, MIX and Dex38.

3T3-F442A cells potentially give rise to at least two adipogenic lineages.
(A). Schematic showing 3T3-F442A cells under the effects of different inducers to express adipogenic phenotypes. (B). Diagram depicting Dex metabolic effects and its relation with the disorders associated with the adipose tissue at organ and systemic level.
In cultures that received St and Dex, the expression of pparg2 decreased during the induction stage (first 4 h) and then increased during the stabilization stage (within the first 8 h of culture). The cells treated with St alone did not exhibit any changes in the expression of pparg2 until after 30 h (the end of the stabilization stage), after which the expression of this gene increased significantly. The decrease in the expression of pparg2 observed during early adipogenesis (induction stage) in the presence of Dex appears to be an effect induced by the glucocorticoid and not an early initiation of the adipose differentiation program. However, the observed expression of this gene during the stabilization stage and the data that have been published by many investigators indicates that this gene is essential for the regulation of the stabilization stage of adipogenesis (not the induction of the program), the transition into adipogenesis and the expression of the adipose phenotype. The expression of cebpa reached the same level in the cultures treated with or without Dex for 30 h (in the stabilization stage). Therefore, Dex modulates the early expression of pparg2 without promoting any early changes in the expression of cebpa, which is the mutually transactivating gene of pparg2. These findings raise the possibility that the early expression of pparg2 in the Dex-treated cultures might cause an early increase in the level of PPARγ2 proteins that are not yet activated by their endogenous ligand, which is a fatty acid derivative39. Thus, the cells must undergo other important changes to initiate the transactivation of the cebpa pathway and establishment of differentiation. This hypothesis is supported by some of our results because we showed that the addition of rosiglitazone, which is a synthetic activator of PPARγ, during the early stages of adipogenesis prompted an earlier expression of cebpa10. Dex appears to stimulate pparg2 expression before the activation of its protein, which is required for the transcription of C/EBPα mRNA. It is remarkable that the expression of these two adipogenic genes in the absence of Dex occurs at similar times during the stabilization stage of adipogenesis.
We also evaluated the expression of srebf1a and srebf1c, which encode the isoforms -1a and -1c of the SREBP1 transcription factor. These proteins are involved in the expression of lipogenic enzymes40 in different cell types and tissues41. Dex did not affect the timing of the expression of these genes and as we showed previously10, these genes play a role during adipogenesis; however, their expression levels are enhanced by the effect of Dex. The -1a and -1c isoforms displayed distinct expression behavior along the differentiation program. Early expression of srebf1a depends on GSK3β activity, through C/EBPβ Thr188 phosphorylation10. Since Dex increased the adipogenic phosphorylation in C/EBPβ Thr188, it could explain the increase in srebf1a expression and the consequent increase in the expression of adipogenic genes pparg2, cebpa and srebf1c with an increase in the number of adipose clusters. On the other hand, expression of srebf1c depends on the transcriptional activity of PPARγ, since activation of this protein by rosiglitazone enhances and accelerates the expression of srebf1c10. The expression levels of both genes, srebf1a and srebf1c, began to increase within the first 4 h of induction, although the expression of the srebf1a gene increased more rapidly than that of srebf1c. In addition, knockdown of srebf1a expression showed that SREPB1a is a key early transcription factor of the adipogenesis program that is observed even earlier than PPARγ210.
In contrast with its effect during the early stages of differentiation, the addition of Dex to mature adipocytes downregulated the expression of the adipogenic genes. This finding can have implications on the action of glucocorticoids in obesity. Previous animal studies have shown that glucocorticoids exert contradictory effects on adipose tissue metabolism42,43, which reflects the difficulty of the in vivo study of adipose metabolism. Therefore, our studies in 3T3-F442A cultures that contain mostly terminally differentiated adipocytes provided results that reflect the effect of Dex in a more homogenous cell population than the in vivo studies. Our results showed that the expression of the lipogenic genes pparg2, cebpa, srebp1c and fabp4 in terminally differentiated adipocytes was reduced by the addition of Dex, which suggests that glucocorticoids impair lipogenesis in adipose cells. In addition, because C/EBPα directly regulates the expression and/or translocation of the glucose transporter GLUT444, the decrease in the expression of cebpa would directly impair the function of GLUT4, which would decrease glucose transport in fat cells, as our data confirmed. Remarkably, Dex also decreased the expression of lep and adipoq to levels similar to those found in non-differentiated 3T3-F442A cells (Figure 5B). Because these adipokines downregulated by Dex are involved in the glucose tolerance signaling pathway, this finding suggests that part of the systemic effect of glucocorticoids involves the development of insulin resistance through alterations in energy metabolism45.
Our data allow us to hypothesize that glucocorticoids might have different simultaneous effects on adipose tissue, as follows. For example, because adipose tissue is composed of adipose progenitor cells and terminally differentiated adipocytes, glucocorticoids might increase the recruitment of the adipose cell progenitors that are induced to differentiate, alter the lipid metabolism through reduced lipogenesis and increased lipolysis and induce the development of insulin resistance in mature adipocytes. It is known that leptin levels controls satiety46, therefore a decrease in the expression of this gene would imply that changes in feeding behavior in animals should take place contributing also to a state of obesity. All these effects ultimately increase the adipose cell number, thereby leading to obesity and induce an unbalance in the lipid metabolism of adipose tissue, which contributes to the development of an insulin-resistant state. Because the accumulation of triglycerides in fat cells is subject to constant turnover, hydrolysis and re-esterification, it is expected that this unbalanced lipid metabolism and lipolysis in the adipose tissue would induce the release of free fatty acids, which are significant contributors to insulin resistance and metabolic syndrome34. It is conceivable that the insulin-resistant state induced by glucocorticoids would be a combination of increased lipolysis and decreased lipogenesis and glucose transport, of which the latter is due to the downregulation of cebpa expression and GLUT4 action.
The other pathway that participates in lipid metabolism is fatty acid oxidation, which, although it was not explored in this work, warrants further investigation. It has been reported that adiponectin increases fatty acid oxidation in muscle47. Therefore, the downregulation of adipoq expression by Dex observed in this study strongly suggests that glucocorticoids would also induce changes in the adiponectin levels of adipocytes that would ultimately impair fatty acid oxidation in other cells.
Methods
Ethics statement
All of the animal experiments were performed according to the NIH guidelines for the welfare of laboratory animals and with the consent of the Internal Committee for the Care and Use of Laboratory Animals (Approved Protocol Number 0051-02. CICUAL, CINVESTAV-IPN).
Materials
Eagle's medium modified by Dulbecco and Vögt (DMEM) and TRIzol® reagent were purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). Adult bovine serum was obtained from HyClone Thermo Fisher Scientific (Waltham, MA, USA). We obtained adult cat serum by bleeding domestic adult cats in observance of the NIH guidelines on the welfare of research animals and using protocols approved by the Internal Committee for the Care and Use of Laboratory Animals of CINVESTAV-IPN. Epidermal growth factor was purchased from Upstate Inc. (Charlottesville, VA, USA). Insulin, d-biotin, human transferrin, triiodothyronine, St, Dex, β-mercaptoethanol, Oil Red O and dimethyl sulfoxide were all obtained from Sigma Chemical Co. (Saint Louis, MO, USA). All other reagents were of analytical grade.
Cell culture
The 3T3-F442A cells (kindly gifted by Dr. Howard Green, Harvard Medical School) were seeded in culture dishes at a cellular density of 1.25 × 103 cell/cm2 in non-adipogenic growing medium, which consisted of DMEM supplemented with 4% adult cat serum, 5 μg/ml insulin and 1 μM d-biotin6. Two days post-confluence, the cultures were transferred to adipogenic medium, which consisted of DMEM supplemented with 2% adult cat serum, 0.2% bovine serum albumin, 5 μg/ml insulin, 5 μg/ml transferrin, 1 μmol/l d-biotin, 2 nmol/l triiodothyronine, 40 μmol/l β-mercaptoethanol, 0.01 ng/ml epidermal growth factor48 and 10 nM St for 4 h as previously described9. The cultures were then transferred to non-adipogenic medium, which was the same as the adipogenic medium with the exception of St unless otherwise stated and allowed to differentiate for 144 h, at which most of the adipogenic cells had differentiated into mature adipocytes.
Evaluation of adipose conversion
At the end of the experiments, the cultures were fixed overnight with 3.4% formalin in PBS at 4°C. The cells were then stained with 0.2% Oil Red O in 60% isopropanol for 2 h at room temperature. The stained dishes were washed with tap water and allowed to air dry49. To quantify the amount of adipogenesis, we manually counted the number of adipose clusters under a stereoscope. The cells that were induced to differentiate during treatment formed adipose clusters9 and since each cluster arises from the selective multiplication of a single parental cell50, the number of adipose clusters corresponds with the number of induced cells.
Analysis of gene expression
The cellular RNA was isolated with TRIzol® reagent from the cell cultures. One microgram of total RNA was transcribed into cDNA using 20 μl of a SuperScript II™ reverse transcriptase reaction mix (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. Relative quantitative PCR was performed through the addition of 1 μl of cDNA to 10 μl of the reaction mix from the FastStart Universal SYBR Green I Master (Rox) kit (Roche Applied Science, Manheim, Germany). The reaction amplification was monitored on a 7500 real-time thermal cycler (Applied Biosystems). The specific primers for each gene are listed in Table 1. The expression values were determined using the 2-ΔΔCT equation and the expression of each gene was normalized to the expression of the ribosomal phosphoprotein large P0 (rplp0) gene that was amplified from the same sample. The changes in gene expression are expressed as relative fold changes.
Immunoblotting
The cultured cells were placed in a 100-mm dish and lysed with 300 μl of the ProteoJET Mammalian Cell Lysis Reagent (Fermentas Inc., Glen Burnie, MA, USA) supplemented with 1X Complete® protease inhibitor (Roche Applied Science, Manheim, Germany), 25 nmol/l sodium pyrophosphate, 10 nmol/l β-glycerophosphate and 10 nmol/l sodium orthovanadate. Thirty micrograms of protein from each sample were separated by 10% SDS-PAGE and blotted for 1 h at 500 mA onto nitrocellulose membranes. The membranes were blocked with 5% non-fat milk in TBS with 0.05% Tween-20 (TBST). The membranes were then challenged with the following antibodies: anti-phospho-Thr188 C/EBPβ (1:1000 dilution; Cell Signaling Technology, Berkeley, CA, USA), anti-C/EBPβ (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or anti-Rab guanine nucleotide dissociation inhibitor (GDI, 1:5000 dilution; Invitrogen, Carlsbad, CA, USA). The membranes were incubated with the primary antibodies overnight at 4°C and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at a 1:10000 dilution for 1 h at room temperature. All of the washes were performed 3 times with TBST for 15 min under constant agitation.
Lipolysis determination
The cultures of terminally differentiated adipocytes were transferred to phenol red-free medium and treated with 0.25 μM Dex in the presence or absence of 1 μM RU486. As a lipolysis control, parallel cultures were treated with 2 mM dibutyryl-cAMP (db-cAMP). After 24 h, the media was removed and the soluble glycerol was determined using the Free Glycerol Determination Kit (Sigma-Aldrich; Saint Louis, MO, USA) according to the manufacturer instructions. The reactions were read at a wavelength of 540 nm. The data are presented as the amount of nmol per protein milligram ± S.D.
Glucose uptake
The 3T3-F442A adipocyte cultures were maintained for 5 days in adipogenic medium with or without 0.25 μM of Dex. The cells were incubated for 60 min in Krebs buffer containing 0.1% bovine serum albumin, 80 μM 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (NBDG) and 100 nM insulin. After the 60-min incubation, the free NBDG was washed from the cultures and the fluorescence associated with the cell monolayers was measured in a Synergy HT fluorescence reader (Bio-Tek Winooski, VT, USA). The results are presented as the mean ± S.D. of 2 independent experiments (n = 16).
Data presentation and statistical analysis
The data presented are the mean ± S.D. of at least 3 independent experiments. The data were analyzed using the non-parametric Mann-Whitney U-test for the comparison of 2 data groups or using the Kruskal-Wallis test for the comparison of 3 or more data groups. A difference was considered statistically significant at a P ≤ 0.05.
References
Green, H. & Kehinde, O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell 5, 19–27 (1975).
Green, H. & Kehinde, O. Formation of normally differentiated subcutaneous fat pads by an established preadipose cell line. J Cell Physiol 101, 169–171 (1979).
Green, H. & Meuth, M. An established pre-adipose cell line and its differentiation in culture. Cell 3, 127–133 (1974).
Mandrup, S., Loftus, T. M., MacDougald, O. A., Kuhajda, F. P. & Lane, M. D. Obese gene expression at in vivo levels by fat pads derived from s.c. implanted 3T3-F442A preadipocytes. Proc Natl Acad Sci U S A 94, 4300–4305 (1997).
Green, H. & Kehinde, O. Spontaneous heritable changes leading to increased adipose conversion in 3T3 cells. Cell 7, 105–113 (1976).
Kuri-Harcuch, W. & Green, H. Adipose conversion of 3T3 cells depends on a serum factor. Proc Natl Acad Sci U S A 75, 6107–6109 (1978).
Gamou, S. & Shimizu, N. Adipocyte differentiation of 3T3-L1 cells in serum-free hormone-supplemented media: effects of insulin and dihydroteleocidin B. Cell Struct Funct 11, 21–30 (1986).
Rubin, C. S., Hirsch, A., Fung, C. & Rosen, O. M. Development of hormone receptors and hormonal responsiveness in vitro. Insulin receptors and insulin sensitivity in the preadipocyte and adipocyte forms of 3T3-L1 cells. J Biol Chem 253, 7570–7578 (1978).
Diaz-Velasquez, C. E., Castro-Munozledo, F. & Kuri-Harcuch, W. Staurosporine rapidly commits 3T3-F442A cells to the formation of adipocytes by activation of GSK-3beta and mobilization of calcium. J Cell Biochem 105, 147–157 (2008).
Ayala-Sumuano, J. T. et al. Srebf1a is a key regulator of transcriptional control for adipogenesis. Sci Rep 1, 178 (2011).
Masuzaki, H. et al. A transgenic model of visceral obesity and the metabolic syndrome. Science 294, 2166–2170 (2001).
Yu, C. Y. et al. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS One 5, e15188 (2010).
Arnaldi, G. et al. Diagnosis and complications of Cushing's syndrome: a consensus statement. J Clin Endocrinol Metab 88, 5593–5602 (2003).
Christ-Crain, M. et al. AMP-activated protein kinase mediates glucocorticoid-induced metabolic changes: a novel mechanism in Cushing's syndrome. FASEB J 22, 1672–1683 (2008).
Colao, A. et al. Persistence of increased cardiovascular risk in patients with Cushing's disease after five years of successful cure. J Clin Endocrinol Metab 84, 2664–2672 (1999).
Hamm, J. K., el Jack, A. K., Pilch, P. F. & Farmer, S. R. Role of PPAR gamma in regulating adipocyte differentiation and insulin-responsive glucose uptake. Ann N Y Acad Sci 892, 134–145 (1999).
Wang, L., Shao, Y. Y. & Ballock, R. T. Peroxisome Proliferator-Activated Receptor-gamma Promotes Adipogenic Changes in Growth Plate Chondrocytes In Vitro. PPAR Res 2006, 67297 (2006).
Yamanouchi, K. et al. Both PPARgamma and C/EBPalpha are sufficient to induce transdifferentiation of goat fetal myoblasts into adipocytes. J Reprod Dev 53, 563–572 (2007).
Cao, Z., Umek, R. M. & McKnight, S. L. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev 5, 1538–1552 (1991).
Kim, J. B. & Spiegelman, B. M. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 10, 1096–1107 (1996).
Tang, Q. Q., Zhang, J. W. & Daniel Lane, M. Sequential gene promoter interactions of C/EBPbeta, C/EBPalpha and PPARgamma during adipogenesis. Biochem Biophys Res Commun 319, 235–239 (2004).
Tang, Q. Q., Otto, T. C. & Lane, M. D. CCAAT/enhancer-binding protein beta is required for mitotic clonal expansion during adipogenesis. Proc Natl Acad Sci U S A 100, 850–855 (2003).
Tang, Q. Q., Otto, T. C. & Lane, M. D. Mitotic clonal expansion: a synchronous process required for adipogenesis. Proc Natl Acad Sci U S A 100, 44–49 (2003).
Tang, Q. Q. et al. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc Natl Acad Sci U S A 102, 9766–9771 (2005).
Bertagna, X., Bertagna, C., Luton, J.-P., Husson, J.-M. & Girard, F. The New Steroid Analog RU 486 Inhibits Glucocorticoid Action in Man. J Clin Endocrinol Metab 59, 25–28 (1984).
Hung, S. H. et al. Pioglitazone and dexamethasone induce adipogenesis in D1 bone marrow stromal cell line, but not through the peroxisome proliferator-activated receptor-gamma pathway. Life Sci 82, 561–569 (2008).
Mendoza-Figueroa, T., Hernandez, A., De Lourdes Lopez, M. & Kuri-Harcuch, W. Intracytoplasmic triglyceride accumulation produced by dexamethasone in adult rat hepatocytes cultivated on 3T3 cells. Toxicology 52, 273–286 (1988).
Rockall, A. G. et al. Hepatic steatosis in Cushing's syndrome: a radiological assessment using computed tomography. Eur J Endocrinol 149, 543–548 (2003).
Bujalska, I. J., Kumar, S. & Stewart, P. M. Does central obesity reflect "Cushing's disease of the omentum"? Lancet 349, 1210–1213 (1997).
Reincke, M. Subclinical Cushing's syndrome. Endocrinol Metab Clin North Am 29, 43–56 (2000).
Wang, F. S., Ko, J. Y., Yeh, D. W., Ke, H. C. & Wu, H. L. Modulation of Dickkopf-1 attenuates glucocorticoid induction of osteoblast apoptosis, adipocytic differentiation and bone mass loss. Endocrinology 149, 1793–1801 (2008).
Masuzaki, H. et al. Glucocorticoid regulation of leptin synthesis and secretion in humans: elevated plasma leptin levels in Cushing's syndrome. J Clin Endocrinol Metab 82, 2542–2547 (1997).
Barahona, M. J. et al. Persistent body fat mass and inflammatory marker increases after long-term cure of Cushing's syndrome. J Clin Endocrinol Metab 94, 3365–3371 (2009).
de Ferranti, S. & Mozaffarian, D. The perfect storm: obesity, adipocyte dysfunction and metabolic consequences. Clin Chem 54, 945–955 (2008).
Skillington, J., Choy, L. & Derynck, R. Bone morphogenetic protein and retinoic acid signaling cooperate to induce osteoblast differentiation of preadipocytes. J Cell Biol 159, 135–146 (2002).
Park, B. H., Qiang, L. & Farmer, S. R. Phosphorylation of C/EBPbeta at a consensus extracellular signal-regulated kinase/glycogen synthase kinase 3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol Cell Biol 24, 8671–8680 (2004).
Gerber, A. N., Masuno, K. & Diamond, M. I. Discovery of selective glucocorticoid receptor modulators by multiplexed reporter screening. Proc Natl Acad Sci U S A 106, 4929–4934 (2009).
Feldman, B. J., Streeper, R. S., Farese Jr, R. V. Yamamoto, K. R. Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects. Proc Natl Acad Sci U S A 103, 15675–15680 (2006).
Kim, J. B., Wright, H. M., Wright, M. & Spiegelman, B. M. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci U S A 95, 4333–4337 (1998).
Amemiya-Kudo, M. et al. Transcriptional activities of nuclear SREBP-1a, -1c and -2 to different target promoters of lipogenic and cholesterogenic genes. J Lipid Res 43, 1220–1235 (2002).
Shimomura, I., Shimano, H., Horton, J. D., Goldstein, J. L. & Brown, M. S. Differential expression of exons 1a and 1c in mRNAs for sterol regulatory element binding protein-1 in human and mouse organs and cultured cells. J Clin Invest 99, 838–845 (1997).
Diamant, S. & Shafrir, E. Modulation of the activity of insulin-dependent enzymes of lipogenesis by glucocorticoids. Eur J Biochem 53, 541–546 (1975).
Niu, C. S., Yeh, C. H., Yeh, M. F. & Cheng, J. T. Increase of adipogenesis by ginsenoside (Rh2) in 3T3-L1 cell via an activation of glucocorticoid receptor. Horm Metab Res 41, 271–276 (2009).
Wu, Z. et al. Cross-Regulation of C/EBPalpha and PPARgamma Controls the Transcriptional Pathway of Adipogenesis and Insulin Sensitivity. Mol Cell 3, 151–158 (1999).
Weyer, C. et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 86, 1930–1935 (2001).
Rentsch, J., Levens, N. & Chiesi, M. Recombinant ob-gene product reduces food intake in fasted mice. Biochem Biophys Res Commun 214, 131–136 (1995).
de Oliveira, C. et al. High-fat diet and glucocorticoid treatment cause hyperglycemia associated with adiponectin receptor alterations. Lipids Health Dis 10, 11 (2011).
Morikawa, M., Green, H. & Lewis, U. J. Activity of human growth hormone and related polypeptides on the adipose conversion of 3T3 cells. Mol Cell Biol 4, 228–231 (1984).
Ramirez-Zacarias, J. L., Castro-Munozledo, F. & Kuri-Harcuch, W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry 97, 493–497 (1992).
Pairault, J. & Green, H. A study of the adipose conversion of suspended 3T3 cells by using glycerophosphate dehydrogenase as differentiation marker. Proc Natl Acad Sci U S A 76, 5138–5142 (1979).
Acknowledgements
We thank Alberto Rodríguez García for the technical assistance and María Elena Rojano Leal for the secretarial assistance.
Author information
Authors and Affiliations
Contributions
J.T.A.S., C.V.V. and W.K.H. contributed for the experimental design and writing manuscript. J.T.A.S. carried out qRT-PCR assays, Western blots, statistical analysis and figure artwork. C.V.V. qRT-PCR assays, NBDG uptake assays. M.M.M. carried out cell culture of agonists-related and NBDG assays. A.B.L. cultured the cells and performed cell cloning and cluster counting experiments. C.H.M. carried out cell culture and lipolysis assay. All authors reviewed manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Financial disclosure: This work was funded by grants PICDS08-8 from Instituto de Ciencia y Tecnologĺa Distrito Federal (ICyT-DF, Mexico) and 104350 from Consejo Nacional de Ciencia y Tecnologĺa (CONACYT,Mexico). The funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors declare no competing interests.
Electronic supplementary material
Supplementary Information
Supplementary Table S1
Rights and permissions
This work is licensed under a Creative Commons Attribution- NonCommercial-ShareALike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Ayala-Sumuano, JT., Velez-delValle, C., Beltrán-Langarica, A. et al. Glucocorticoid Paradoxically Recruits Adipose Progenitors and Impairs Lipid Homeostasis and Glucose Transport in Mature Adipocytes. Sci Rep 3, 2573 (2013). https://doi.org/10.1038/srep02573
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep02573
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
