
Abstract
Vacuolar-type H+ ATPases (V-ATPases) are multimeric protein complexes that play a universal role in the acidification of intracellular compartments in eukaryotic cells. We have isolated the recessive medaka mutation tintachina (tch), which carries an inactivating modification of the conserved glycine residue (G75R) of the proton pump subunit atp6v1Ba/vatB1. Mutant embryos show penetrant pigmentation defects, massive brain apoptosis and lethality before hatching. Strikingly, an equivalent mutation in atp6v1B1 (G78R) has been reported in a family of patients suffering from distal renal tubular acidosis (dRTA), a hereditary disease that causes metabolic acidosis due to impaired kidney function. This poses the question as to how molecularly identical mutations result in markedly different phenotypes in two vertebrate species. Our work offers an explanation for this phenomenon. We propose that, after successive rounds of whole-genome duplication, the emergence of paralogous copies allowed the divergence of the atp6v1B cis-regulatory control in different vertebrate groups.
Similar content being viewed by others
Introduction
V-ATPases are highly conserved ATP-dependent proton pumps that play a universal role as pH regulators in intracellular acidic organelles of eukaryotic cells. They form multi-subunit complexes assembled in two subdomains: the Vo membrane domain, which is responsible for proton transport across membranes and is comprised of the subunits a, d, c, c″ and e; and the V1 cytoplasmic domain, which is responsible for ATP hydrolysis and is made up of 8 subunit types, designated as A to H. Intracellular V-ATPases play essential roles in receptor-mediated endocytosis, vesicular trafficking between organelles, membrane fusion, protein degradation and autophagy1,2,3. In addition to their role in intracellular compartments, V-ATPases can also pump protons across the plasma membrane, thus acidifying the extracellular medium. This membrane localization has been described in various mammalian cell types including macrophages, osteoclasts and renal intercalated cells. The targeting of V-ATPases to the cell surface is largely mediated by tissue specific a-subunit isoforms. These include a1, a3 and a4, which have been reported at the plasma membrane in neurons, osteoclasts and renal intercalated cells respectively4. In addition, the B subunit kidney-specific isoform (atp6v1B1) has been shown to interact with the PDZ protein NHERF1 through its C-terminal motif5, which in turn regulates the docking of the V-ATPase complex to the apical membrane in renal cells. Interestingly, hereditary diseases linked to mutations in V-ATPase subunits have been reported specifically for these targeting isoforms. Mutations in the osteoclast-specific a3 isoform cause autosomal recessive osteopetrosis6, whereas mutations in both the renal-specific a4 and B1 isoforms cause distal renal tubular acidosis7,8. Similarly, interference with normal proton secretion in the kidney has been observed in atp6v1B1−/−mice9.
Within the vacuolar proton pump, B subunits are essential components of the central A3B3 hexameric barrel of the V1 domain, which is directly responsible for ATP hydrolysis10. While a single atp6v1B gene can be identified in invertebrate genomes11, two different paralogous genes have been reported in mammals: atp6v1B1, which is restricted to a number of tissues including kidney, lung, inner ear, olfactory epithelium and epididymal cells and atp6v1B2, which is broadly expressed, if not practically ubiquitous7,12,13,14. Two different B genes, vatB1 and vatB2, also named atp6v1Ba and atp6v1B2 [ZFIN]15, have also been reported in teleost fish16. It has been postulated that they are, respectively, orthologs of the tetrapods atp6v1B1 and atp6v1B217. So far, no mutations have been described for these subunits in teleosts.
In this study we describe the identification, cloning and analysis of the medaka (Oryzias latipes) mutant tintachina (tch), which carries a loss-of-function mutation in atp6v1Ba/vatB1. The mutation causes pigmentation defects and brain degeneration and involves the missense modification (G75R) of a glycine residue, conserved across eukaryotes, to arginine. An equivalent homozygous mutation in atp6v1B1 (G78R) has been reported in a family affected with dRTA18. This serendipitous coincidence allows comparison of the physiological consequences of an equivalent loss of function in genes that share a common ancestor. Here we analyze the phylogenetic relationship between the different vertebrate B subunits and offer a hypothesis on their evolutionary history and their divergent functional adaptations.
Results
The tintachina mutation disrupts atp6v1Ba/vatB1
In an ENU (N-ethyl-N-nitrosourea) mutagenesis screen for mutations affecting retina development in medaka19 we identified the mutation tintachina (tch), which displayed reduced pigmentation of the eye and punctate body pigmentation (Fig. 1a–d). The mutation was named tintachina (‘chinese ink’ in Spanish) after the characteristic melanocyte pattern and has been transmitted through more than 12 generations without noticeable phenotypic changes. It is a lethal recessive mutation that shows full penetrance and minimal phenotypic variability. The mutant phenotype first becomes apparent as pigmentation emerges between stages 28–29 by reduced pigmentation of the eyes (Figure 1e, f). At early organogenesis, no morphogenetic defects are observed in tch embryos, which show normal organization of body plan and axon scaffolds, as assessed by anti-acetylated tubulin labeling (Figure 1g, h). At later stages, mutant embryos suffer progressive tissue degeneration, particularly in the CNS and finally die between stages 37 to 39, shortly before hatching.

Tch phenotype and positional cloning.
(a–d) Comparison of wt (a,c) and tch (b,d) medaka embryos at stage 35 shows reduced pigmentation in the mutants. (e–f) Mutant phenotype becomes first apparent at stage 29. Red arrows and asterisks indicate hypopigmented eyes and characteristic dotted melanocytes respectively. (g–h) Acetylated tubulin staining of wt (g) and tch (h) stage 33 embryos reveals normal axon scaffold organization (arrows). (i) Genetic and physical map of the medaka tch locus on chromosome 15. Recombinants are indicated in green above the chromosome line. Genetic (cM) and physical (Kb) distances are indicated below. Transcription units are depicted as arrows. Chromosomal markers used for fine mapping are indicated in blue. The genomic structure of atp6v1Ba is depicted: exons are represented as red bars. The mutated exon 3 is indicated with an asterisk. (j) The sequencing trace data from wild-type and tch mutants cDNA and their predicted translation is depicted. Sequencing reveals a G to A point mutation (arrows).
We mapped the tch locus to chromosome 15 by bulk segregation analysis20. Further mapping reduced the region of interest to an interval of 700 kbp, as defined by two flanking restriction length polymorphisms (RFLPs), which contained a few candidate genes including atp6v1Ba/vatB1 (Figure 1i). The characteristic tch phenotype: hypopigmentation of the eyes, punctate melanocytes and progressive brain degeneration, has been described in a number of zebrafish mutants affecting different subunits of the vacuolar proton pump including atp6v0d1, atp6v0c, atp6v1H, atp6v1F and atp6v1E121,22,23. Therefore, we decided to investigate whether the tch mutation was associated with atp6v1Ba/vatB1. A RFLP analysis of the atp6v1Ba/vatB1 locus showed no recombinant chromosomes (0/576) in the mutant embryos (Figure 1i), thus suggesting that it was the mutated gene. To confirm this, the entire coding region of atp6v1Ba/vatB1 was amplified by PCR from cDNA and sequenced in several independent wild type and tch embryos. A missense mutation altering glycine to arginine at position 75 (G75R) was consistently identified in mutant embryos (Figure 1j). This missense mutation was further confirmed by sequencing the genomic region encompassing exon 3 in wild type and tch embryos. The G75R point mutation (-RS-G/R-QVLE-) lies within a highly conserved domain (Supplementary Figure S1) in a glycine residue preserved in all metazoans and even in other eukaryotes, such as the yeast Saccharomyces. A L81P point mutation (-RSGQV-L/P-E-) in the same conserved domain of atp6v1B1 has been identified as causative for dRTA in humans7. Moreover, an equivalent homozygous mutation (G78R: -RS-G/R-QVLE-) caused by the same nucleotide substitution (g/a) has been reported in a Turkish family affected with dRTA18.
Lysosomal function and neuronal survival are compromised in tch
Oculocutaneous albinism is a common trait in congenital disorders affecting different aspects of pigment cell biology. Anomalies range from melanophore specification/migration defects to abnormal melanosome maturation and melanin synthesis. To analyze at which level tch embryos are affected, we examined the mutants in the background of the transgenic line Tyr::GFP, which labels differentiated melanophores and to a lesser extent xanthophores24. As a parallel control we crossed the viable albino mutation heino25, which also affects melanin synthesis, into the same Tyr::GFP line. A normal distribution and number of GFP-positive cells were observed in tch and heino (Figure 2a–c), thus indicating that melanophore migration and differentiation is unaffected in both mutants. This observation is in line with previous reports showing that V-ATPase function is required for both melanosome maturation and melanin synthesis22. In addition, the orange auto-fluorescent sepiapterins, synthesized in xantophores in acidic organelles homologous to the melanosomes24, appear to be completely absent in tch (Figure 2c). This suggests a more general requirement for atp6v1Ba/vatB1 in the acidification and biogenesis of lysosomal-related acidic organelles. To further investigate this, we labeled live embryos with LysoTracker Green, a probe that selectively accumulates in acidic intracellular compartments. After a 10 min pulse, LysoTracker accumulation was imaged in the dorsal diencephalon of stage 31 tch embryos and wild type siblings. Whereas the probe was significantly incorporated into acidic compartments in wild type cells, a very limited accumulation was observed in tch (Figure 2d, e), thus indicating that lysosomal acidification is compromised in the mutants.

Lysosomal function and neuronal survival are compromised in tch.
(a–c) Melanophores, as revealed by the transgenic line Tyr::GFP (green) and auto-fluorescent xantophores (orange) are shown in wild type (a), heino (b) and tch (c) embryos at stage 31. Eye pigmentation defects (asterisk) are shown in fluorescent and transmitted light images (insets). White arrow in “c” points to dotted melanocytes in tch. (d–e) LysoTracker Green in vivo labelling reveals acidic organelles (arrows) at the dorsal diencephalon (see inset in d) in wild type (d) and tch (e) stage 31 embryos. Tyr::GFP positive melanophores (m) are indicated with dotted lines as an internal reference. (f–i) Tunel staining shows apoptotic cells in wt (f,g) and tch (h,i) medaka embryos at stage 33 in the anterior brain (f,h) dorsal neural tube and fin folds (g,i).
Previous reports have shown that lysosomal dysfunction mediated by V-ATPase inhibition induces apoptosis in mammalian cells26,27. Conversely, the overexpression of atp6v1B2 in HEK cells increased their resistance to apoptosis28. To further understand the progressive degeneration observed in tch embryos, we investigated by TUNEL assay whether this was due to an enhanced apoptotic rate. Whole mount TUNEL staining in stage 33 embryos revealed increased apoptosis in tch. In the mutants, apoptotic cells were particularly detected in the retina, diencephalon and mesencephalon (Figure 2 f–h) and to a lesser extent in the dorsal neural tube, ventral mesoderm and fin folds (Figure 2g–i). Therefore, we confirmed a role for atp6v1Ba/vatB1 as a regulator of lysosomal function and neuronal survival.
The evolution of V-ATPase B subunits within the vertebrate group
It has been postulated that atp6v1Ba/vatB1 is the true teleost ortholog of the mammalian gene atp6v1B117. To confirm this point and to further investigate the evolutionary history of the atp6v1B family, we identified and analyzed atp6v1B genes in vertebrate and invertebrate genomes. Multiple sequence alignments revealed that vertebrate B subunits are highly conserved, showing an average % of aminoacid identity of 86.9±0.4 (n = 20; mean±SEM) when compared to Drosophila atp6V1B (Supplementary Figure S1). We then generated a protein family tree using a maximum likelihood approach (Figure 3a). In contrast to what would be expected for orthologous genes, teleost atp6v1Ba/vatB1 and mammalian atp6v1B1 proteins did not cluster together in the tree. In fact, phylogenetic analyses defined three different B subunit monophyletic groups: an atp6v1B2 group that includes B2 subunits from all studied jawed vertebrate lineages (average % of identity with Drosophila 87.8±0.7); an atp6v1Ba group that includes teleost proteins (average % of identity with Drosophila 87.1±0.1) and finally a somewhat more divergent atp6v1B1 group comprising of tetrapod and coelacanth proteins (average % of identity with Drosophila 85.1±0.5).

The evolution of V-ATPase B subunits within the vertebrate group.
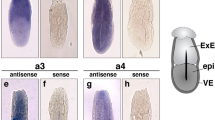
(a) Phylogenetic analyses defined three different B subunits groups (here color-coded). The syntenic arrangement of the different genes is indicated. See also Supplementary Figure S1 and Table 1. (b) Schematic representation of the evolutionary history of atp6v1B genes in vertebrates. Whole genome duplications and atp6v1B gene loss events at the different lineages under the most parsimonious scenario are depicted. See also Supplementary Figures S3 and S4. (c–g) Atp6v1Ba expression in medaka embryos revealed by whole-mount ISH at stages 25 (c), 28 (d, f) and 33 (e, g). Arrows point to specific expression in the diencephalon (c–e), dorsal neural tube (f) and pronephros (g). (h–j) Atp6V1BaP4Kb::GFP expression in stage 33 embryos. Arrows point to specific expression in the nervous system (h, i) and the pronephros (j).
Teleost genomes share an extra third round of whole genome duplication (or 3R) that occurred at the root of the lineage29 and may obscure orthology assignment when genes in tetrapods and teleosts are compared. To investigate this possibility, we studied the genome of the spotted gar (Lepisosteous oculatus), a basal ray-finned fish that diverged from teleosts before the occurrence of the 3R event30,31. Importantly, we found gar representatives for each of the three paralogs: Ba, B1 and B2 (Figure 3a). This suggests that Ba genes are not divergent duplicated teleost orthologs of any of the tetrapod genes, but ancient jawed vertebrate paralogs that emerged after the first two rounds of genome duplication (2R) that happened during the evolution of vertebrates32,33. Subsequently, B1 and Ba would have been selectively lost in all the studied species of the teleost and sarcopterigian (tetrapods and coelacanth) lineages respectively (Figure 3b). In fact, divergent Ba pseudogenized remnants are still detectable in sarcopterigian species such as coleacanths, painted turtles and humans (Supplementary Figure S2). Therefore, our phylogenetic analyses indicate that the medaka atp6v1Ba/vatB1 protein belongs to the atp6v1Ba clade (Figure 3a). Consistently, in situ hybridization analysis of atp6v1Ba expression in medaka embryos (Figure 3c–g) showed a very similar pattern to that reported for the zebrafish atp6v1Ba ortholog in the database ZFIN15. In early organogenesis stages, atp6v1Ba could be detected at low levels in most medaka tissues (Figure 3c). As development proceeds, atp6v1Ba transcripts accumulation was progressively detected in the central nervous system; particularly in the diencephalon, retina and dorsal neural tube (Figure 3c–f). An additional expression domain was also detected in the anterior pronephros at stage 33 (Figure 3g). Atp6v1Ba complex expression is in contrasts with the more restricted atp6v1B1 expression in mammals. In fact, it rather resembles atp6v1B2 expression, which was initially described as an isoform ubiquitously expressed at low levels in a range of human tissues, but at significantly higher levels in brain and kidney12.
Syntenic analysis of the atp6v1B gene family
To complement our phylogenetic studies we examined synteny conservation between vertebrate genomes around the atp6v1B paralogous loci and between the equivalent regions in two non-vertebrate deuterostomes: the basal chordate amphioxus (Branchiostoma floridae) and the hemichordate Saccoglossus kowalevskii (Supplementary Figure S3). Extensive conservation across species in this genomic neighborhood further demonstrated the orthology relationships previously uncovered by the phylogenetic analysis, fully supporting the existence of three duplicated atp6v1B genes in the last common ancestor of bony vertebrates. As in the case of the B subunits, many of the genes located in their vicinity (e. g. Shootin, Slc18a and Gfra) were also present in several copies in most of the studied genomes. Consistent with their origin at the vertebrate whole genome duplication events, the location of most of these duplicates is confined to the same set of paralogous chromosomal segments where the atp6v1B genes are located: 10q26, 2p13 and 8p22 in the human genome (Supplementary Figure S3). Furthermore, some of the copies were located in an additional chromosomal segment, 5q31-33 in humans, raising to four the number of paralogous chromosomes derived from the same ancestral linkage group, in full agreement with the general pattern of quadruple conserved macrosynteny typical of vertebrate genomes33. This implies that a fourth atp6v1B gene was present in the ancestral jawed vertebrate after the 2R events and was subsequently lost before bony vertebrate radiation.
We then compared the syntenic arrangement of the three paralogous regions (Ba, B1 and B2) containing atp6v1B genes plus the fourth region, currently devoid of an atp6v1B copy (hereafter B0), to infer their evolutionary relationships. Three different topologies are possible: [Ba-B1, B2-B0], [Ba-B2, B1-B0] and [Ba-B0, B1-B2]. Among these, the [Ba-B0, B1-B2] grouping was not consistent with any differentially shared syntenic arrangement, as it would imply more gene losses than the other two scenarios. The other two topologies, [Ba-B1, B2-B0] and [Ba-B2, B1-B0], were both supported by the presence of differentially shared duplicated genes (Supplementary Figure S4). Although the arrangement [Ba-B1, B2-B0] may seem somewhat more likely, any hypothesis should be interpreted with caution as gene loss is a pervasive phenomenon in evolution, especially in the context of the genetic redundancy created by whole genome duplications34. In either case, both topologies indicate that atp6v1Ba and atp6v1B1 are paralogous genes radiating from a common ancestor that lay in close proximity to a vax gene (Supplementary Figure S4).
Cis-regulatory sub-functionalization within the atp6v1B gene family
In vertebrates, sub-functionalization of paralogs after gene duplication has frequently occurred by loss or modification of cis-regulatory modules35. The differential expression pattern observed between atp6v1Ba and atp6v1B1 indicates that such a process may have taken place, resulting in divergent, tissue specific, functions. Through transgenesis in mice, it has been shown that a 6.5 kb fragment of the atp6v1B1 promoter (comprising the entire intergenic region between atp6v1B1 and vax2) contains enough regulatory information to recapitulate the specific expression of the gene in the kidney, lung and epididymis14. Interestingly, this intergenic region between atp6v1B1 and vax2 comprises of only a few kb in tetrapods, while the equivalent region between atp6v1Ba and vax1 expands over 10–20 kb in more compact fish genomes (Table 1). This suggests that 5′ cis-regulatory elements, responsible for the widespread expression of the gene, may have been lost at the atp6v1B1 locus after genome duplication. According to this hypothesis, such elements would have been retained in the intergenic region between atp6v1Ba and vax1. To test this possibility we analyzed the regulatory information contained in the 5′ region of the medaka atp6V1Ba. A 4 kb fragment upstream of the gene was fused to GFP and injected into medaka embryos to generate stable reporter lines (Atp6V1BaP4Kb::GFP). The expression pattern driven by this element was already visible in F0 embryos and was further consistently confirmed in F1 for three independent insertions. In agreement with atp6V1Ba expression, GFP signal was weakly detected at early stages in medaka embryos (not shown) and increased later in development, accumulating mainly in the central nervous system and pronephros at stage 33 (Figure 3h–j). This finding indicates that the intergenic region between atp6v1Ba and vax1 contains sufficient cis-regulatory information to recapitulate the widespread and likely ancestral, expression of the gene.
Discussion
Here we report the identification and analysis of the mutation tch, a missense mutation (G75R) of the proton pump subunit atp6v1Ba that causes oculocutaneous albinism, progressive brain degeneration and embryonic lethality. Our analyses indicate that atp6v1Ba plays a role in the acidification of intracellular compartments and hence its lack of function induces apoptosis in those tissues in which it is normally expressed. Consistent with the essential role of V-ATPase complexes in organelle acidification, mutations in almost any of its subunits cause embryonic lethality11,22,23. Exceptions to this are those subunits involved in intracellular targeting, such as the mammalian atp6v1B1, which shows a tissue-restricted distribution and does not play an essential role during embryogenesis9. In this study we have presented evidence showing that the teleost gene atp6v1Ba/vatB1 is not the real ortholog of the mammalian gene atp6v1B1, as previously postulated17; on the contrary, we show that atp6v1Ba and atp6v1B1 are paralogous genes which emerged after whole genome duplication events. Despite the fact that both proteins share high sequence similarity (>80% of identity) and play a conserved molecular function, their mutations result in fundamentally different phenotypes. Although subtle changes in protein sequence could partially explain this discrepancy, it is likely that atp6v1Ba and atp6v1B1 divergent expression patterns have a major impact in their physiological role, thus accounting for a large part of the phenotypic variation observed when mutated. In line with this, our data suggest that gene duplication within the vertebrate atp6v1B family has allowed the divergence of their cis-regulatory modules. This process may have relieved evolutionary constraints, allowing atp6v1B1 to acquire tissue-restricted expression. In turn, specific expression would facilitate the adaptation of the protein from a lysosomal/endosomal function to a role at the plasma membrane. At the physiological level, the membrane targeting of the B1 subunit represented an important adaptation in the context of the functional requirements of the mammalian kidney4 or the amphibian larval skin36.
Methods
Fish stocks and transgenic lines
Medaka (Oryzias latipes) strains Cab and Kaga were kept as described previously19. The Kaga strain was used for chromosomal assignation and positional cloning. The stable Tyr::eGFP line has been previously described37. To generate the stable atp6V1Baprom4Kb::GFP line using I-SceI mediated transgenesis, a fragment of 4 Kb upstream of the atg of the medaka atp6V1ba gene was amplified and fused to eGFP in a meganuclease compatible vector38. The coordinates of this fragment in the medaka genome are the following: Ch15: 3952227-3956219.
Genetic mapping and positional cloning
tch was assigned to chromosome 15 by bulk segregation analysis using the Kaga strain as a reference20. The genetic distance to the locus was narrowed by available genetic markers39 on 576 mutant chromosomes. We used the markers Ola1002d (8/240; 3.3 cM) and MF01SSA078A08 (3/384; 0.78 cM) as initial references flanking the mutation. From these anchoring positions, new restriction length polymorphisms were designed and examined as nested reference points through the chromosome. The amplified medaka atp6v1Ba full-length sequence and the 3′ terminal fragment (377 bp) of the medaka atp6v1B2 have been deposited in GeneBank (JX416286 and JX416287 respectively).
LysoTracker staining
Embryos were dechorionated with Proteinase K (10 mg/ml in H2O) and hatching enzyme as described37. Stage 31 dechorionated embryos were incubated for a 10 min pulse in 1 μM LysoTracker Green (DND26; Invitrogene) at 25°C and then imaged by confocal microscopy using a Leica SP5 system.
Whole-mount acetylated tubulin staining
were performed as follows: embryos were fixed with 20% DMSO/80% MeOH at room temperature for 2 hours, treated with 10% H2O2/10% DMSO/80% MeOH overnight at 4°C and incubated with anti-acetylated tubulin (Sigma T6793) monoclonal antibodies at 1:1000 dilution. After incubation with peroxidase-coupled secondary anti-mouse antibodies (1:2000 Sigma) samples were stained using a NovaRed substrate kit (Vector Laboratories).
Whole-mount in situ hybridization
Whole-mount in situ was performed using digoxigenin labeled riboprobes as described37.
Whole mount tunel staining
Embryos were with fixed with 4% p-formaldehyde in PBS overnight at 4°C, stored in 100% methanol and then rehydrated in PBS-tween 0.1% (PBT) before treatment with proteinase k (0.2 mg/ml in PBT) for one hour. After post-fixation with 4% p-formaldehyde apoptotic cells were identified using the in situ cell death detection kit AP (Roche). Alkaline phosphatase associated signal was visualized with FastRed (Roche).
Search for atp6v1B genes, phylogenetic analysis and genome browsing
In species where atp6v1B genes were not previously identified or available, or where gene predictions were fragmentary or poorly annotated, we built new manually curated predictions as described previously40. In the case of medaka and Tetraodon atp6v1B2 genes some gaps/Ns (indicated by “X” in Supplementary Figure 1) have to be included in order to maintain an ancestral exon/intron boundary and the reading frame near the translation start codon, respectively. This suggests that these genes could be subjected to an ongoing pseudogenization process. We aligned atp6v1B proteins using MAFFT41 as implemented in Jalview 2.842 and discarded poor quality regions (the N- and C- terminal portions, the first 29 and the last 23 positions in the alignment of Supplementary Figure 1) from the alignment for subsequent phylogenetic analysis. We built maximum likelihood trees with MEGA 543, under the most complex available model (WAG + I + Γ) and 100 bootstrap replicates. Very divergent sequences, such as those of fly, sea squirt and frog atp6v1B2 were excluded from phylogenetic analysis.
Genomes of the studied species were browsed through the JGI (http://genome.jgi-psf.org/euk_home.html), NCBI (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi), UCSC (http://genome.ucsc.edu/) and Ensembl (http://www.ensembl.org/info/about/species.html) webpages, using the following genome versions: Branchiostoma floridae v1.0, Chrysemys picta v3.0.1, Ciona intestinalis v2.0, Danio rerio Zv9, Drosophila melanogaster R5.48, Gasterosteus aculeatus 1.0, Homo sapiens Build 37, Latimeria chalumnae v1, Lepisosteus oculatus v1, Mus musculus Build 38, Oryzias latipes 2.0, Petromyzon marinus v7.0, Saccoglossus kowalevskii Build1.1, Tetraodon nigroviridis v8 and Xenopus tropicalis v4.1. Eel atp6v1B gene complement was inferred from the Japanese eel (Anguilla japonica) genome downloaded from the eel genome website (www.eelgenome.com/). For the alignment and phylogenetic analyses, full length protein sequences of the European eel (A. anguilla) atp6v1Ba and atp6v1B2 orthologs (previously published under accession numbers AAD55091 and AAC78641, respectively) were used.
References
Jefferies, K. C., Cipriano, D. J. & Forgac, M. Function, structure and regulation of the vacuolar (H+)-ATPases. Arch Biochem Biophys 476, 33–42 (2008).
Mijaljica, D., Prescott, M. & Devenish, R. J. V-ATPase engagement in autophagic processes. Autophagy 7, 666–8 (2011).
Nishi, T. & Forgac, M. The vacuolar (H+)-ATPases—nature's most versatile proton pumps. Nat Rev Mol Cell Biol 3, 94–103 (2002).
Wagner, C. A. et al. Renal vacuolar H+-ATPase. Physiol Rev 84, 1263–314 (2004).
Breton, S. et al. The B1 subunit of the H+ATPase is a PDZ domain-binding protein. Colocalization with NHE-RF in renal B-intercalated cells. J Biol Chem 275, 18219–24 (2000).
Frattini, A. et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat Genet 25, 343–6 (2000).
Karet, F. E. et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21, 84–90 (1999).
Smith, A. N. et al. Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat Genet 26, 71–5 (2000).
Finberg, K. E. et al. The B1-subunit of the H(+) ATPase is required for maximal urinary acidification. Proc Natl Acad Sci U S A 102, 13616–21 (2005).
Vasilyeva, E., Liu, Q., MacLeod, K. J., Baleja, J. D. & Forgac, M. Cysteine scanning mutagenesis of the noncatalytic nucleotide binding site of the yeast V-ATPase. J Biol Chem 275, 255–60 (2000).
Davies, S. A. et al. Analysis and inactivation of vha55, the gene encoding the vacuolar ATPase B-subunit in Drosophila melanogaster reveals a larval lethal phenotype. J Biol Chem 271, 30677–84 (1996).
van Hille, B. et al. Heterogeneity of vacuolar H(+)-ATPase: differential expression of two human subunit B isoforms. Biochem J 303 (Pt 1), 191–8 (1994).
Paunescu, T. G., Jones, A. C., Tyszkowski, R. & Brown, D. V-ATPase expression in the mouse olfactory epithelium. Am J Physiol Cell Physiol 295, C923–30 (2008).
Miller, R. L. et al. V-ATPase B1-subunit promoter drives expression of EGFP in intercalated cells of kidney, clear cells of epididymis and airway cells of lung in transgenic mice. Am J Physiol Cell Physiol 288, C1134–44 (2005).
Sprague, J. et al. The Zebrafish Information Network: the zebrafish model organism database. Nucleic Acids Res 34, D581–5 (2006).
Boesch, S. T., Eller, B. & Pelster, B. Expression of two isoforms of the vacuolar-type ATPase subunit B in the zebrafish Danio rerio. J Exp Biol 206, 1907–15 (2003).
Niederstatter, H. & Pelster, B. Expression of two vacuolar-type ATPase B subunit isoforms in swimbladder gas gland cells of the European eel: nucleotide sequences and deduced amino acid sequences. Biochim Biophys Acta 1491, 133–42 (2000).
Borthwick, K. J. et al. A phenocopy of CAII deficiency: a novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J Med Genet 40, 115–21 (2003).
Loosli, F. et al. Mutations affecting retina development in Medaka. Mech Dev 121, 703–14 (2004).
Martinez-Morales, J.-R., Naruse, K., Mitani, H., Shima, A. & Wittbrodt, J. Rapid chromosomal assignment of Medaka mutants by bulked segregant analysis. Gene 329, 159–165 (2004).
Golling, G. et al. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet 31, 135–40 (2002).
Madsen, E. C. & Gitlin, J. D. Zebrafish mutants calamity and catastrophe define critical pathways of gene-nutrient interactions in developmental copper metabolism. PLoS Genet 4, e1000261 (2008).
Nuckels, R. J., Ng, A., Darland, T. & Gross, J. M. The vacuolar-ATPase complex regulates retinoblast proliferation and survival, photoreceptor morphogenesis and pigmentation in the zebrafish eye. Invest Ophthalmol Vis Sci 50, 893–905 (2009).
Yamamoto, T. Medaka (Killifish), Biology and Strains, (Keigaku Publishing Company, Tokyo, 1975).
Loosli, F. et al. A genetic screen for mutations affecting embryonic development in medaka fish (Oryzias latipes). Mechanisms of Development 97, 133–139 (2000).
Nakashima, S. et al. Vacuolar H+-ATPase inhibitor induces apoptosis via lysosomal dysfunction in the human gastric cancer cell line MKN-1. J Biochem 134, 359–64 (2003).
De Milito, A. et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res 67, 5408–17 (2007).
Li, G. et al. A novel cellular survival factor--the B2 subunit of vacuolar H+-ATPase inhibits apoptosis. Cell Death Differ 13, 2109–17 (2006).
Amores, A. et al. Zebrafish hox clusters and vertebrate genome evolution. Science 282, 1711–4 (1998).
Amores, A., Catchen, J., Ferrara, A., Fontenot, Q. & Postlethwait, J. H. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics 188, 799–808 (2011).
Hoegg, S., Brinkmann, H., Taylor, J. S. & Meyer, A. Phylogenetic timing of the fish-specific genome duplication correlates with the diversification of teleost fish. J Mol Evol 59, 190–203 (2004).
Dehal, P. & Boore, J. L. Two rounds of whole genome duplication in the ancestral vertebrate. PLoS Biol 3, e314 (2005).
Putnam, N. H. et al. The amphioxus genome and the evolution of the chordate karyotype. Nature 453, 1064–71 (2008).
Maeso, I., Roy, S. W. & Irimia, M. Widespread recurrent evolution of genomic features. Genome Biol Evol 4, 486–500 (2012).
Woolfe, A. & Elgar, G. Comparative genomics using Fugu reveals insights into regulatory subfunctionalization. Genome Biol 8, R53 (2007).
Quigley, I. K., Stubbs, J. L. & Kintner, C. Specification of ion transport cells in the Xenopus larval skin. Development 138, 705–14 (2011).
Martinez-Morales, J. R. et al. ojoplano-mediated basal constriction is essential for optic cup morphogenesis. Development 136, 2165–75 (2009).
Thermes, V. et al. I-SceI meganuclease mediates highly efficient transgenesis in fish. Mech Dev 118, 91–8 (2002).
Naruse, K. et al. A detailed linkage map of medaka, Oryzias latipes: comparative genomics and genome evolution. Genetics 154, 1773–1784 (2000).
D'Aniello, S. et al. Gene expansion and retention leads to a diverse tyrosine kinase superfamily in amphioxus. Mol Biol Evol 25, 1841–54 (2008).
Katoh, K., Kuma, K., Toh, H. & Miyata, T. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res 33, 511–8 (2005).
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–91 (2009).
Tamura, K. et al. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol Biol Evol 28, 2731–9 (2011).
Acknowledgements
We thank Felix Loosli, Lazaro Centanin and Mariana Delfino-Machin for their critical input and Rocío Polvillo for their excellent technical assistance. We thank Nathan Kenny for proofreading the manuscript. We also thank Kyle Martin for help with the analysis of the Japanese eel genome. IM holds a posdoctoral contract supported by the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) / ERC grant [268513]. This work was supported by grants BFU2008-04362, BFU2011-22916 and P11-CVI-7256 to JRMM.
Author information
Authors and Affiliations
Contributions
Most of the experimental work was done by C.M. and J.R.M.-M. in J.W.'s and J.R.M.-M.'s laboratories. I.M. contributed with key phylogenetic analyses. The manuscript was written by J.R.M.-M.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by/3.0/
About this article
Cite this article
Müller, C., Maeso, I., Wittbrodt, J. et al. The medaka mutation tintachina sheds light on the evolution of V-ATPase B subunits in vertebrates. Sci Rep 3, 3217 (2013). https://doi.org/10.1038/srep03217
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03217
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
