
Abstract
Structural analysis of branched DNA molecules (BDM) is important as model systems for DNA junctions and also as building units for DNA assembly. Although there have been efforts to study the structures of BDM, label-free solution structures have not been well determined yet. Here, we used a combination of synchrotron-based experimental tools and computational simulation to study the global structures of label-free BDM in solution. Overall structures of 3-arm and 4-arm BDM were revealed as an asymmetric T(or Y)-shape and a distorted X-shape, respectively. The internal structures of the DNA double helix were shown to have a canonical B-form for both the BDM. We also reconstructed the thermal denaturation process of BDM by determining the transient global structures over a wide range of temperatures. The proposed high-resolution structures of BDM are expected to provide fundamental information for studies of the biological function of junction DNAs and DNA assembly.
Similar content being viewed by others

Introduction
Branched DNA molecules (BDM) are widely employed models for studying DNA junctions, which are central intermediates in important biological processes, such as genome duplication, DNA damage repair and homologous recombination of DNA1,2. In addition to these biological applications, BDM have also been used more recently, with the development of DNA technology, as important building blocks in the construction of complicated DNA nanostructures via molecular self-assembly3,4,5,6,7,8. Because the increased importance of BDM are due to their structural significance, many studies have aimed to determine the structures of BDM using techniques such as atomic force microscopy9, fluorescence resonance energy transfer (FRET)10,11,12 and X-ray crystallography13,14. Despite these efforts, however, determining high-resolution, three-dimensional (3D) structures in aqueous solution, where DNA molecules are dispersed, remains a challenge due to technical limitations15,16,17. Sample immobilization, trapping, chemical labeling, or crystallization, which are required for using these structural tools, prohibit the discovery of realistic structures of branched DNA. Structures determined by these methods allow only limited information because of a lack of freedom in configurations of DNA molecules, especially in the aqueous phase.
Solution X-ray scattering is a powerful method to determine solution-phase, label-free structures of macromolecules. The structure determination based on the assumption of single molecular envelop has become increasingly convenient owing to the compilation of shape determination tools18,19. In addition, DNA assembly is generally performed in solution phase. However, due to the intrinsically reduced information, which is one-dimensional and orientationally averaged scattering intensity profile, difficulties still remain in probing systems with conformational flexibility and multiplicity20. To address this point, ensemble-based reconstruction methods have been suggested that offer insight into the conformational transition21,22,23,24.
In this report, we show that the combination of synchrotron small-angle X-ray scattering (SAXS), wide-angle X-ray diffraction (WAXD) and atomistic simulations can be powerful in structural studies of label-free, BDM dispersed in aqueous solution. By using synthetic 3-arm and 4-arm BDM as model systems for three-way junctions (3 WJ) and four way junctions (4 WJ), respectively, we have reconstructed the global structures of junction DNAs in the aqueous phase. In addition to determining high-resolution structures at room temperature, we have also determined the transient global structures of BDM over a wide range of temperatures to study the denaturation process of 3 WJ and 4 WJ DNA.
Results
In order to synthesize monodisperse BDM, we employed the one-pot synthesis method7,25: equal moles of all 3 and 4 oligonucleotides were mixed together to form the 3-arm and 4-arm BDM (hereafter, referred to as 3 WJ and 4 WJ DNA, respectively). The formation of BDM was evaluated by gel electrophoresis (Supplementary Fig. S1), in which the mobility of DNA molecules depends on their size, shape and extent of base pairing. The estimated yield of branched DNA is close to 100%. The sequences of the oligonucleotides are presented in Supplementary Table S1.
Determination of 3 WJ and 4 WJ DNA structures in aqueous buffer at room temperature by solution SAXS
Synchrotron SAXS measurements of 3 WJ and 4 WJ DNA in PBS buffer were carried out at 25°C (Fig. 1). Guinier analysis26, performed on the SAXS data (Supplementary Fig. S2), revealed linearity in a small q region which suggests that the DNA molecules exist in a monodisperse conformational state at each temperature. In Figure 1a, we display SAXS intensity curves, I(q), measured for 3 WJ and 4 WJ DNA at 25°C. To provide an intuition for overall molecular shape in real space, the pair-distance distributions, p(r), are shown in Figure 1b, which were calculated by the indirect Fourier transform of the entire scattering profiles using the program GNOM27 To further facilitate direct structural comparison, we created atomistic structures of 3 WJ and 4 WJ DNA by molecular dynamic (MD) simulations. The aforementioned quantities, I(q) and p(r), can be computed from the atomic coordinates as well, through the use of the program CRYSOL28 and GNOM27, respectively. The calculated SAXS curves and pair-distance distributions for representative configurations of 3 WJ and 4 WJ DNA obtained from MD simulations are generally in agreement with the experimental results at 25°C, as shown in Figure 1a and 1b. For a quantitative comparison for p(r), we obtain the radius of gyration, Rg,p(r) and the maximum dimension, Dmax, as the distance where the p(r) function approaches zero. The results for Rg,p(r) and Dmax obtained from experiment at room temperature agree reasonably with those from the simulation results, as listed in Supplementary Table S2. We observe that both the experimental and simulation results of p(r) exhibit three main peaks characterizing geometry of BDM in Figure 1b. The first peak at r ≈ 2 nm reflects average distances between backbones, corresponding to the average diameter of double helix. The second one around r ≈ 3.5 nm, although not distinct, attributes to atom pairs constituting the pitch of helix turn. The third peak located at r ≈ 5 nm corresponds to average distance between each helix arms, which determines the overall size of molecular envelope.

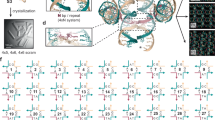
SAXS data and structural models.
(a) Scattering profiles of 3 WJ and 4 WJ in PBS buffer at 25°C. Insets show 2D SAXS patterns. The symbols are the experimental data and the solid lines are SAXS profiles obtained from the reconstructed structural models using the ab initio shape determination program DAMMIF. The dashed lines without symbols are the theoretical SAXS curves of structural models calculated from MD simulations. For clarity, each plot is shifted along the log I axis. (b) Pair distance distribution functions for 3 WJ and 4 WJ, based on an analysis of the experimental SAXS data using the program GNOM. The dotted lines without symbols are the p(r) functions for structural models obtained from MD simulations labeled as dashed lines in a. (c–d) Reconstructed structural models of 3 WJ and 4 WJ in PBS buffer with and without any symmetry restrictions from the ab initio shape determination program DAMMIF DAMMIF (Chi = 1.599 (P1), 1.603 (P2), 1.611 (P3) for 3 WJ and Chi = 1.545 (P1), 1.549 (P2), 1.544 (P4) for 4 WJ). (e–f) Structural models calculated from MD simulations for 3 WJ and 4 WJ using simulation employing the parmbsc0 force field. Surface rendering in the structural models was achieved using Discovery Studio 1.6 (Accelrys Inc.).
To corroborate the p(r) function analysis and to obtain a visual 3D representation of the structure of 3 WJ and 4 WJ DNA in different conformational states, we reconstructed model-independent structures that occupy a search volume defined by the maximum dimension, Dmax, calculated from the p(r) function using the ab initio shape determination program DAMMIF29, an upgraded version of DAMMIN30. Multiple runs of DAMMIF were used to generate structural models, with and without imposing symmetry constraints (No symmetry, P2 symmetry, P3 symmetry and P4 symmetry). The structural models for 3 WJ DNA in PBS buffer at 25°C exhibited distorted Y-shaped forms composed of 3 arms and the junction in the innermost center of the reconstructed models, except in that with imposed P3 symmetry restriction (Fig. 1c). The structural models for 4 WJ DNA in PBS buffer at 25°C adopted distorted X shaped forms consisting of 4 arms and the junction near the center of the reconstructed models, which slightly deviated from an ideal X-shaped conformation with imposing P4 symmetry constraint. Some structural differences in the overall shape were observed depending on the type of symmetry restriction imposed (Fig. 1d). The reconstructed conformations resemble the atomic structures obtained from MD simulation for both 3 WJ and 4 WJ DNA in Figures 1e and 1f. It is fascinating that 3D structures of DNA molecules in solution were successfully determined from X-ray scattering experiments without any constraint such as labeling or trapping.
A detailed description of equilibrium structures obtained from MD simulations is provided in Supplementary Information (Figs. S3 and S4 and Table S3). In particular, we carefully compared the results obtained from 2 force field parameters that have been widely used. We also discuss the discrepancies between the experimental results and simulation results in the p(r) patterns in detail, by means of the relationship between the general features in the p(r) function and the overall geometry of the DNA molecules (Figs. S5 and S6).
General features of branched DNA by solution WAXD
With the aim of better understanding the helical structures within DNA molecules, synchrotron WAXD measurements of 3 WJ and 4 WJ DNA in PBS buffer were performed at 25°C (Fig. 2). To gain further insight into the internal helical structures of DNA molecules, we evaluated ensemble-averaged WAXD patterns of the 2,500 model configurations calculated from MD simulations using the program CRYSOL. The calculated WAXD patterns can be compared with the experimental WAXD patterns at 25°C by means of the positions and intensities of diffraction peaks, labeled P1–P6 in Figure 2. Scattering from structural repeats in helical DNA molecules produces a series of diffraction peaks in the WAXD pattern that vary in position and intensity, depending on the molecular structure31,32. Figure 2 shows ensemble-averaged WAXD patterns of full (structure 3 in Fig. 2) and partial (structures 4 and 5 in Fig. 2) structures of the 2,500 model configurations of a 3 WJ DNA molecule calculated from MD simulations. Scattering caused by the stacked nucleotide bases can be estimated from the partial structure 4 in Figure 2, which yields a single diffraction peak, P6, shown by the orange line. Peaks in the small angle region, P1–P4, originate from scattering due to the sugar-phosphate backbone, as shown by the green line in the WAXD pattern calculated from the partial structure 5 in Figure 2. However, we do not observe a diffraction peak, P5, that arise predominately from cross-terms between the backbone and stacked base pairs which are enforce in a B′-form DNA conformation32. As shown in Figure 2, the experimental WAXD patterns for both 3 WJ and 4 WJ DNA molecules in PBS buffer at 25°C exhibited P1–P6 diffraction peaks, with the exception of P5, that are characteristic of a typical B-form DNA conformation.

WAXD profiles from experimental studies and MD simulation.
WAXD fingerprint patterns from 3 WJ (blue) and 4 WJ (red) in PBS buffer at 25°C. Ensemble-averaged WAXD patterns calculated from MD simulations for the complete structure of 3 WJ (black), the base pair atoms only (orange) and the sugar-phosphate backbone atoms only (green).
Conformational change of branched DNA in thermal denaturation
To fully understand the conformational behavior of DNA, it is necessary to define the structure of both the native state and the various denatured states with the change of environments. Recent studies have revealed the biological significance of denatured states, functional flexibility and intrinsic disorder that occurs in functional regions and interfaces under physiologically active conditions22,33. However, the detailed structural features of BDM and the variations in structure that occur in response to temperature change have not been well studied.
To gain a visual 3D representation of the overall structures and the structural differences of 3 WJ and 4 WJ DNA molecules in solution at various temperatures, we attempted to reconstruct structural models using the ab initio shape determination program DAMMIF. The experimental SAXS curves and their p(r) functions at temperature range between 25°C and 72°C are shown in Figure 3. As shown in Figures 3a and 3b, the scattering profiles changed in the wide q region (q > 1.5 nm−1) between 63°C and 69°C. The scattering patterns at 72°C were clearly changed in the whole q region, due to conformational transition from the branched DNA duplex structure to a fully random coil conformation of single-stranded DNA. These results were consistent with the disappearance of the 3rd peak, which is characteristic of a branched DNA form, in the p(r) function (Supplementary Table S2). The structural parameters obtained from the SAXS data of 3 WJ and 4 WJ DNA molecules in solution at various temperatures are listed in Supplementary Table S2. On the whole, the obtained values of both DNA molecules at 72°C were markedly decreased in comparison with those measured at 25–69°C. From these results, we observed that both 3 WJ and 4 WJ DNA underwent a substantial conformational change at 72°C during the thermal denaturation process. Such observations which occurred in the thermal process could be explained in detail by the 3D reconstructed models. Figure 3e, f show the structural models reconstructed for 3 WJ and 4 WJ DNA molecules in PBS buffer at various temperatures using DAMMIF. The reconstructed models were obtained with and without imposing any restrictions on the symmetry of the DNA molecule. As shown in Figure 3, structural differences in the overall shape were apparent depending on the symmetry restriction, but all models are very meaningful for structure interpretation. However, in order to strike a balance between symmetry underestimate from P1 (no symmetry) constraint and symmetry overestimate from P3 (for 3 WJ) and P4 (for 4 WJ) constraints, we focused primarily on the structural models reconstructed with imposing P2 symmetry restriction that consistently represent the most reasonable and adequate feature for a systematic understanding of the conformational change in response to change in temperature. As already shown, the structural models reconstructed for 3 WJ DNA in solution at 25°C include 3 distinct arms of the same length. However, in the 3 WJ DNA structures modeled at 40–63°C, under early stages of the denaturation process, 2 of the 3 arms were somewhat shortened compared with those of the 3 WJ DNA at 25°C. These results suggest that the 3 WJ DNA underwent a slight conformational change during thermal denaturation, which is rearrangement of the denatured strands that was caused by breakdown of hydrogen bonding between bases within individual arm domains. In addition, it can be predicted that at least 1 arm domain is likely to retain a certain degree of structural stability in the overall shape under the present conditions. The structural models reconstructed for 3 WJ DNA in solution at both 66 and 69°C revealed bent forms that were different from those of 3 WJ DNA in solution at lower temperature; one of the 3 arms completely disappeared due to destruction of the DNA duplex structure. These results seem to be correlated to the change of ratio of the 1st peak intensity values to the 3rd peak ones in the p(r) function. We observed that the contribution of the 3rd peak was gradually decreased as the thermal denaturation was proceeding. The 3 WJ DNA molecule in solution at 72°C, during the final stages of denaturation, adopted a fully random coil conformation of single-stranded DNA, which is confirmed by analysis of p(r) for single-stranded DNA obtained from MD simulation (Supplementary Figs. S7 and S8 and Table S4). The 4 WJ DNA in solution at 25°C was found to exist in a distorted X-shaped conformation containing four arms of the same size and was very similar to the simulated conformation. The structural models reconstructed for 4 WJ DNA in solution at 40 to 60°C, in the early stages of denaturation, have similar four-armed conformations overall to those of 4 WJ DNA at 25°C, but more distorted X-shaped forms. Interestingly, from the 4 WJ DNA structures modeled at around 63°C, we found that the 4 WJ DNA experienced a considerable structural change, where the molecule changed from a 4-armed to a 3-armed conformation. The structural models reconstructed for 4 WJ DNA maintained their structure based on the 3-armed conformation up to 69°C. These results suggest the possibility that one or two of the 4 arms are denatured in advance and then they are aggregated to form a partially molten globule. Interestingly, structural models (at 63 to 69°C) reconstructed by imposing a P4 symmetry constraint have some similarities to those (3-armed conformation) reconstructed based on P2 symmetry restriction, while retaining P4 symmetry characteristics in the right model of each column. The 4 WJ DNA molecule in solution at 72°C, during final stages of thermal denaturation, adopted a fully random coil conformation of single stranded DNA, similar to the 3 WJ DNA in solution at 72°C. From these reconstructions, we were able to identify an intermediate DNA conformation that was dominant at each temperature in the thermal denaturation process.

Structural models in response to change in temperature based on small-angle X-ray scattering.
Scattering profiles of 3 WJ (a) and 4 WJ (b) in PBS buffer as a function of temperature. The symbols are the experimental data and the solid lines are SAXS profiles obtained from reconstructed structural models by using the ab initio shape determination program DAMMIF. For clarity, each plot is shifted along the log I axis. Pair distance distribution functions for 3 WJ (c) and 4 WJ (d) in solutions of various temperatures, based on an analysis of the experimental SAXS data using the program GNOM. Reconstructed structural models of 3 WJ (e) and 4 WJ (f) in PBS buffer, with and without any symmetry restrictions, as a function of temperature using the ab initio shape determination program DAMMIF (Chi = 1.477 ~ 1.635 for 3 WJ and Chi = 1.447 ~ 1.789 for 4 WJ). Surface rendering in the structural models was achieved using Discovery Studio 1.6 (Accelrys Inc.).
In order to understand these thermal denaturation process, we additionally performed ultra violet (UV) absorbance and circular dichroism (CD) spectra analysis. The thermal denaturation of double stranded DNA is progressive and this can be seen in a sigmoidal increasing of UV absorption. By increasing temperature, the transition from the double stranded to denatured structure was observed over the range of approximately 60 to 70°C. The Tm values determined from UV absorption spectra are 64 and 63°C for 3 WJ and 4 WJ, respectively (Fig. S9). After 72°C, both BDMs are fully denatured to three and four single stranded oligonucleotides. These thermal behaviors were also shown from the CD spectroscopic analysis. (Fig. S10). These UV and CD results are consistently well matched with previously described SAXS characterization.
MD simulation results at high temperatures
We have performed MD simulation of the atomistic model of BDM at elevated temperature in order to obtain a microscopic description of structural transitions that occur during thermal denaturation. Our model system for BDM solvated in PBS buffer remains stable rigidly for longer than 500 ns at 400 K (Figs. S11 and S12). In order to facilitate denaturation, we increased the temperature of the system equilibrated at 400 K to 450 K instantly, which eventually causes melting within 100 ns. We note that the temperature applied in our model system does not scale consistently with the physical temperature. The use of unphysical temperature was intended to apply an enhanced noise level to the system. We discuss the simulation result at high temperatures in Supplementary Information in detail.
In Figure 4, the melting process is depicted with isosurface plots for the probability density of finding atoms in 3D space, computed from three subsequent trajectories of 30 ns for 3 WJ and 4 WJ systems. In the experiment, averaging over numerous molecules and time is reflected in the diffraction pattern, such that mobile portions produce relatively low intensity. Likewise, portions of molecules that are relatively unstable should yield a lower probability density of atom existence in our analysis. In this manner, the isosurface plot is considered a reasonable counterpart of MD simulation to understand reconstructed structures from the experiment. In Figure 4, we additionally show the probability density of finding Na+ ions. Specifically, the isosurfaces correspond to the position where the chance of finding DNA and Na+ ions is 30% and 1%, respectively. Pair-distance distributions, p(r), computed from instantaneous structures are also presented indicating partially disordered structures of 3 WJ and 4 WJ DNA in Figure 4. While the molecules undergo thermal denaturation, changed are the positions of the peaks in p(r) and size of the molecules. In describing the melting process, we focused on 3 features that are common to both 3 WJ and 4 WJ. After increasing the temperature, enhanced thermal noise causes large fluctuations of both local and global structures, including inter-helix angles. As a result, a distorted structure of the backbone facilitates base opening for 0–30 ns. Interestingly, base pairs of 2 helices start to flip out, while those of the other helix remain intact. Next, for 30–60 ns, the 2 helices with broken hydrogen bonds are shortened to form a partially molten globule. Finally, after 60 ns, 2 helices are aggregated, leading to the complete collapse of 1 helix. This aggregation is not considered the result of any targeted interaction, such as the formation of hydrogen bonds between 2 helices, but the cooperative effect of surrounding counterions and water molecules. The attractive electrostatic interaction between negatively charged phosphate and Na+ ions compete with the hydration force exerted on Na+ ions34. With the increased thermal noise, the hydration effect of water molecules for both the DNA coil and ions is reduced, resulting in a higher probability that Na+ ions will be located near DNA, as shown in Figure 4. This observation provides a microscopic picture of reconstructed model structures presented in Figure 3, especially for shortened or missing helix at the intermediate stages of denaturation.

Global structures of 3 WJ and 4 WJ derived from MD simulations.
Three stages of melting processes in 3 WJ and 4 WJ systems, obtained from nonequilibrium MD trajectories, where the temperature is increased from 400 K to 450 K instantly at t = 0. (a) Probability isosurfaces where the chance of finding atoms for 3 WJ DNA (blue) and Na+ ions (magenta), computed with 3 subsequent trajectory segments of 30 ns long, are 30% and 1%, respectively. (b) Probability isosurface for 4 WJ DNA using the same criteria as in a. In each, the right panels are pair-distance distributions, p(r), for individual configuration of 3 WJ and 4 WJ at each stage.
Discussion
In summary, we show the entire process of synthesizing and structurally analyzing solvated 3 WJ and 4 WJ by combining SAXS, WAXD experiments and atomistic simulations over a wide range of the temperature. The global structures of BDM are determined successfully by interpreting the SAXS intensity profiles through the use of shape determination software. Revealed structures are an asymmetric T- (or Y-) shape and a distorted X-shape for 3 WJ and 4 WJ DNA, respectively, at room temperature. We also determined from the WAXD experiments that the double helix structures in both branched DNAs belong to a canonical B-form. Moreover, our results provide an intermediate DNA conformation, which was dominant at each temperature during thermal denaturation, by analyzing the results from in-situ temperature SAXS in solution and MD simulations. In the latter, we offer a microscopic insight for thermal denaturation of BDM, taking the cooperative dynamics of counterions and water molecules into consideration. The methods applied in this article could be used for studying structures of other nucleic acids, such as DNA hairpins, enzymes, bent sequences and RNA, as well as proteins and protein complexes. In general, solution SAXS can provide solution structural information at resolutions often sufficient for functional insights into how these bio molecules work in the context of conformational and assembly state under physiological conditions. The results reported here will guide the development of new DNA nanostructural and self-assembly techniques using BDM as building units.
Methods
Sample preparation
The sequences of oligonucleotides for BDM were modified from Luo's X- and Y-DNA25,35. Oligonucleotides were resuspended in TE buffer. The 3 WJ and 4 WJ DNA were constructed by mixing three or four 36-bp oligonucleotides, respectively, in PBS buffer. 3 WJ and 4 WJ DNA were synthesized and purified following the methods described previously7,25,35.
X-ray data acquisition and processing
SAXS measurements were carried out using the 4 C SAXS II beamline of the Pohang Light Source II (PLS II) with 3 GeV power at the Pohang University of Science and Technology (POSTECH), Korea. A light source from an In-vacuum Undulator 20 (IVU 20: 1.4 m length, 20 mm period) of the PLS II storage ring was focused with a vertical focusing toroidal mirror coated with rhodium and monochromatized with a Si (111) double crystal monochromator, yielding an X-ray beam wavelength of 0.675 Å. The X-ray beam size at the sample stage was 0.2 (V) × 0.6 (H) mm2. A two-dimensional (2D) charge-coupled detector (Mar USA, Inc.) was employed. A sample-to-detector distance of 2.00 m for SAXS and 0.25 m for WAXD were used. The magnitude of scattering vectors, q = (4π/λ) sin θ, was 0.2 nm−1 < q < 25 nm−1, where 2θ is the scattering angle and λ is the wavelength of the X-ray beam source. The scattering angle was calibrated with polyethylene-b-polybutadiene-b-polystyrene block copolymer and silver behenate standards. We used solution sample cells with 10 μm thick mica windows, a volume of 50 μL and an X-ray beam path length of 0.8 mm. All scattering measurements were carried out at various temperatures by using a JULABO FP50-HL control system with a temperature stability of ± 0.01°C. The SAXS data were collected in 5 successive frames of 0.1 min each to monitor radiation damage. There were no changes in the scattering patterns with time, i.e., no radiation damage was detected during the scattering measurements. To obtain high-quality scattering data without any interferences of DNA molecules (ie., concentration effects) (Supplementary Table S2), measurements of DNA solutions were carried out over a small range of oligonucleotide concentrations (3.2–6.7 mg/mL). In addition, the molecular mass of each sample has been calculated to confirm the consistency between the expected molecular masses from the sequence of the nucleotides and those calculated from the forward scattering I(0) (in the absolute scale after normalization against concentration and water scattering) from Guinier extrapolation (Supplementary Table S5). Each 2D SAXS/WAXD pattern was circularly averaged from the beam center and normalized to the transmitted X-ray beam intensity, which was monitored with a scintillation counter placed behind the sample. The scattering of the PBS buffer solution was used as the experimental background.
MD simulation and structural analyses
We built atomistic structures of 3 WJ and 4 WJ DNA, corresponding to the sequences displayed in Supplementary Table S1, using the NAB tool included in the AMBERtools12 suite36. Specifically, the structures of double helices for each arm were constructed separately in a generic B-form using NAB and arranged appropriately to connect the inter-helical bonds and form the Y-shape and X-shape of 3 WJ and 4 WJ, respectively. We then minimized the molecule for 2 ps using the steepest descent algorithm and raised the temperature to 300 K gradually over 100 ps to relax the inter-helical bonds, which were initially very awkward, while monitoring the bond potential energy. Although not perfectly relaxed, the resulting structures of DNA molecules were found to be stable throughout the subsequent procedures of solvation and equilibration.
Most of the present MD simulations were performed using the GROMACS software package37 with the all-atom parmbsc0 force fields38, which is known to preserve the stable double helix structure during long simulations39. For comparison, we also carried out simulations using the parm99 force field40. The DNA molecule was neutralized with 0.15 M Na+ and Cl− ions and solvated with more than 28000 TIP3P water molecules41 in a rectangular box. The size of the simulation cell was set to ensure a solvation shell of 10 Å thickness around the DNA molecule. We carried out MD simulations at constant temperature (300–450 K) and pressure (1 bar) using the Berendsen algorithm42, with a time constant of 1.0 ps for heat bath coupling and pressure relaxation. The compressibility was set to 4 × 10−5 bar−1, corresponding to the water system. An integration time step of 1 fs was selected and the electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method43 with a real space cut-off of 9 Å. Initially, the DNA molecule was relaxed again through the energy minimization with water molecules and ions and allowed to equilibrate at 300 K at constant volume for 500 ps. Subsequently, the equilibration was continued at constant pressure for at least 100 ns, while fluctuations of the root-mean square deviation were monitored. The trajectories obtained from production runs thereafter were used for analyses.
References
Heller, R. C. & Marians, K. J. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 7, 932–943 (2006).
Atkinson, J. & McGlynn, P. Replication fork reversal and the maintenance of genome stability. Nucleic Acids Res. 37, 3475–3492 (2009).
Park, N., Um, S. H., Funabashi, H., Xu, J. & Luo, D. A cell-free protein-producing gel. Nat. Mater. 8, 432–437 (2009).
Pinheiro, A. V., Han, D., Shih, W. M. & Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nature. Nanotech. 6, 763–772 (2011).
Hur, J. et al. DNA hydrogel-based supercapacitors operating in physiological fluids. Sci. Rep. 3, 1282, 10.1038/srep01282 (2013).
Seeman, N. C. DNA in a material world. Nature 421, 427–431 (2003).
Li, Y. et al. Controlled assembly of dendrimer-like DNA. Nature Mater. 3, 38–42 (2004).
Li, Y., Cu, Y. T. H. & Luo, D. Multiplexed detection of pathogen DNA with DNA-based fluorescence nanobarcodes. Nat. Biotechnol. 22, 885–889 (2005).
Shlyakhtenko, L. S., Potaman, V. N., Sinden, R. R., Gall, A. A. & Lyubchenko, Y. L. Structure and dynamics of three-way DNA junctions: atomic force microscopy studies. Nucleic Acids Res. 28, 3472–3477 (2000).
Hohng, S. et al. Fluorescence-force spectroscopy maps two-dimensional reaction landscape of the holliday junction. Science 318, 279–283 (2007).
Sabir, T., Schröder, G. F., Toulmin, A., McGlynn, P. & Magennis, S. W. Global Structure of Forked DNA in Solution Revealed by High-Resolution Single-Molecule FRET. J. Am. Chem. Soc. 133, 1188–1191 (2011).
Sabir, T. et al. Branchpoint Expansion in a Fully Complementary Three-Way DNA Junction. J. Am. Chem. Soc. 134, 6280–6285 (2012).
Ortiz-Lombardía, M. et al. Crystal structure of a DNA Holliday junction. Nat. Struct. Mol. Biol. 6, 913–917 (1999).
Zheng, J. et al. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nature 461, 74–77 (2009).
Young, M. A. et al. Structure determination and analysis of local bending in an A-tract DNA duplex: Comparison of results from crystallography, nuclear magnetic resonance and molecular dynamics simulation on d(CGCAAAAATGCG). Methods Enzymol. 261, 121–128 (1995).
Dixit, S. B., Pitici, F. & Beveridge, D. L. Structure and axis curvature in two dA6 × dT6 DNA oligonucleotides: comparison of molecular dynamics simulations with results from crystallography and NMR spectroscopy. Biopolymers 75, 468–479 (2004).
Wright, P. E. & Dyson, H. J. Linking folding and binding. Curr. Opin. Struct. Biol. 19, 31–38 (2009).
Rambo, R. P. & Tainer, J. A. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA and protein assemblies by small-angle X-ray scattering. Curr. Opin. Struct. Biol. 20, 128–137 (2010).
Petoukhov, M. V. & Svergun, D. I. Applications of small-angle X-ray scattering to biomacromolecular solutions. Int. J. Biochem. Cell Biol. 45, 429–437 (2012).
Oroguchi, T., Hashimoto, H., Shimizu, T., Sato, M. & Ikeguchi, M. Intrinsic Dynamics of Restriction Endonuclease EcoO109I Studied by Molecular Dynamics Simulations and X-Ray Scattering Data Analysis. Biophys. J. 96, 2808–2822 (2009).
Bernadó, P., Mylonas, E., Petoukhov, M. V., Blackledge, M. & Svergun, D. I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 129, 5656–5664 (2007).
Pelikan, M., Hura, G. L. & Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 28, 174–189 (2009).
Yang, S., Blachowicz, L., Makowski, L. & Roux, B. Multidomain assembled states of Hck tyrosine kinase in solution. Proc. Natl. Acad. Sci. USA 107, 15757–15762 (2010).
Różycki, B., Kim, Y. C. & Hummer, G. SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions. Structure 19, 109–116 (2011).
Park, N. et al. High-yield cell-free protein production from P-gel. Nature Protocols 4, 1759–1770 (2009).
Glatter, O. & Kratky, O. Data treatment In Small Angle X-Ray Scattering. 119–196 (London; New York: Academic Press, 1982).
Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 25, 495–503 (1992).
Svergun, D. I., Barberato, C. & Koch, M. H. J. CRYSOL - a Program to Evaluate X-ray Solution Scattering of Biological Macromolecules from Atomic Coordinates. J. Appl. Cryst. 28, 768–773 (1995).
Franke, D. & Svergun, D. I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 42, 342–346 (2009).
Svergun, D. I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 (1999).
Müller, J. J. Calculation of Scattering Curves for Macromolecules in Solution and Comparison with Results of Methods using Effective Atomic Scattering Factors. J. Appl. Cryst. 16, 74–82 (1983).
Zuo, X. et al. X-ray diffraction "fingerprinting" of DNA structure in solution for quantitative evaluation of molecular dynamics simulation. Proc. Natl. Acad. Sci. USA 103, 3534–3539 (2006).
Radivojac, P. et al. Intrinsic Disorder and Functional Proteomics. Biophys. J. 92, 1439–1456 (2007).
Pal, S., Maiti, P. K., Bagchi, B. & Hynes, J. T. Multiple time scales in salvation dynamics of DNA in aqueous solution: the role of water, counterions and cross-correlations. J. Phys. Chem. B 110, 26396–26402 (2006).
Um, S. H. et al. Enzyme-catalysed assembly of DNA hydrogel. Nature Mater. 5, 797–801 (2006).
Case, D. A. et al. AMBER 12. (University of California, San Francisco, 2012).
van der Spoel, D. et al. Gromacs User Manual. 4.5.4 edn, (2010).
Pérez, A. et al. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 92, 3817–3829 (2007).
Orozco, M., Noy, A. & Perez, A. Recent advances in the study of nucleic acid flexibility by molecular dynamics. Curr. Opin. Struct. Biol. 18, 185–193 (2008).
Cheatham III, T. E., Cieplak, P. & Kollman, P. A. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16, 845–862 (1999).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Berendsen, H. J. C., Postma, J. P. M., DiNola, A. & Haak, J. R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Darden, T., York, D. & Pedersen, L. G. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Acknowledgements
The synchrotron X-ray scattering measurements at the Pohang Accelerator Laboratory were supported by the Ministry of Education Science and Technology (MEST) (Basic Science Research Project No. 20090072614).
Author information
Authors and Affiliations
Contributions
K.I., K.S.J. and N.P. designed the experiments. K.I., J.H., K.S.J. and N.P. performed the experiments and analyzed the data. D.J., S.K. performed modeling work. K.I., D.J., K.S.J. and N.P. wrote the manuscript. S.H., K.K. provided advice on the project.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Im, K., Jeong, D., Hur, J. et al. Robust analysis of synthetic label-free DNA junctions in solution by X-ray scattering and molecular simulation. Sci Rep 3, 3226 (2013). https://doi.org/10.1038/srep03226
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep03226
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
