
Abstract
Reconstitution of signaling pathways involving single mammalian transmembrane receptors has not been accomplished in yeast cells. In this study, intact EGF receptor (EGFR) and a cell wall-anchored form of EGF were co-expressed on the yeast cell surface, which led to autophosphorylation of the EGFR in an EGF-dependent autocrine manner. After changing from EGF to a conformationally constrained peptide library, cells were fluorescently labeled with an anti-phospho-EGFR antibody. Each cell was subjected to an automated single-cell analysis and isolation system that analyzed the fluorescent intensity of each cell and automatically retrieved each cell with the highest fluorescence. In ~3.2 × 106 peptide library, we isolated six novel peptides with agonistic activity of the EGFR in human squamous carcinoma A431 cells. The combination of yeast cells expressing mammalian receptors, a cell wall-anchored peptide library and an automated single-cell analysis and isolation system might facilitate a rational approach for de novo drug screening.
Similar content being viewed by others
Introduction
Sensing of extracellular ligands (e.g., hormones, cytokines and growth factors) via receptors on the cell surface modulates intracellular signaling molecules and subsequently regulates gene expression. Analysis of receptor-mediated signal transduction allows delineation of signaling pathways involved in the onset of various diseases. Thus, it is important to find or design drugs with agonistic or antagonistic activity for target receptors.
To date, seven transmembrane G-protein-coupled receptors (GPCRs) have been recognized as potential target molecules for drug discovery (e.g., propranolol as an antagonist of β-adrenergic receptors, phenylephrine as an agonist of α1-adrenergic receptor)1,2. Single transmembrane receptor tyrosine kinases (RTKs) are also potential therapeutic targets. After conventional screening for drugs that bind to RTKs on mammalian cells, molecular target drugs have been developed to modulate these receptors, particularly for the purpose of treating cancers (e.g., gefitinib as an antagonist of the epidermal growth factor receptor (EGFR)3, trastuzumab as an antagonist of HER2 (a member of EGFR family)4). For drug discovery, it is indispensable to prepare a tremendous number of chemical compounds (104 ~ 105 species) to identify lead compounds that target receptors. Because the initial screening is usually undertaken by in vitro administration of each chemical compound to mammalian cells (e.g., Chinese hamster ovary cells over-expressing target receptors), it is laborious to evaluate compounds using a large number of cell cultures. Furthermore, endogenous regulation mechanisms of target receptors, such as redundancy, crosstalk and feedback5,6,7, may affect quantitative measurement of second messenger levels8,9,10. These situations led us to develop a new high-throughput screening system of drugs for target receptors without any interference from endogenous regulation mechanisms.
Because Saccharomyces cerevisiae expresses two endogenous GPCRs, STE2 and STE311, various mammalian GPCRs have been successfully expressed in S. cerevisiae (e.g., adrenergic, chemoattractant C5a, serotonin and somatostatin receptors)12. Some mammalian GPCRs activate the yeast pheromone-sensing pathway in a dose-dependent manner of each ligand13,14, suggesting that yeast is suitable for quantitative analyses of mammalian GPCRs and their ligands. For example, Klein et al.15 succeeded in de novo screening of agonists for the human formyl peptide receptor like-1 receptor (FPRL-1R) using yeast cells co-expressing FPRL-1R and a random peptide library. Yeast cells are also considered as suitable host cells for screening agonists and antagonists of RTKs, because there is no tyrosine kinase activity in yeast cells16. When a constitutively active EGFR is expressed on the yeast cell surface, the EGFR autophosphorylates and recruits Grb2 and Sos near the yeast plasma membrane, followed by activation of the Ras signaling pathway17. However, EGF-dependent activation of the EGFR (i.e., autophosphorylation) has not been detected in yeast cells, probably because the yeast cell wall prevents EGF from accessing the ligand-binding domain of the EGFR.
Here, we report that a cell wall-anchored form of EGF can access the EGFR expressed on the yeast cell surface and EGF activates the EGFR in an autocrine manner. Furthermore, a cell wall-anchored form of a conformationally constrained peptide library18, instead of EGF, allowed us to develop a de novo high-throughput screening system of agonists for the EGFR. In this system, yeast cells were stained by indirect immunofluorescence with an anti-phospho-EGFR antibody and then analyzed with an automated single-cell analysis and isolation system19. Finally, in the ~3.2 × 106 peptide library, we have identified six novel peptides that show agonistic activity of the EGFR in mammalian cells for a short period.
Results
EGF-dependent autophosphorylation of the EGFR in yeast cells
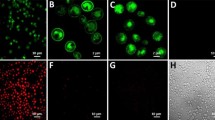
Because the yeast cell wall prevents extracellular macromolecules from accessing the plasma membrane13,14,17, both EGF and the EGFR were concurrently displayed on the yeast cell surface to increase the local concentration of EGF for reconstitution of the EGF signaling pathway in yeast cells (Figure 1a). An N-terminal HA-epitope-tagged human EGF was expressed in the periplasm with the assistance of MFα1-prepro peptide20 and FLO42 (cell wall-anchoring domain of FLO121) (Figure 1b, top). A C-terminal V5-epitope-tagged human EGFR was expressed as a membrane-anchored form with the assistance of SUC2-signal peptide22 (Figure 1b, bottom). Western blot analysis showed that HA-EGF-FLO42 and EGFR-V5 were localized in cell wall and cytosolic fractions, respectively (Figure 1c). Because the calculated molecular weight of mature HA-EGF-FLO42 was 11,783, the faint 12 kDa band and broad >20 kDa bands (left panel) were considered as non-glycosylated and glycosylated forms of HA-EGF-FLO42, respectively. In addition, the calculated molecular weight of mature EGFR-V5 was 133,901 and the broad 130 and 160 kDa bands (right panel) might have corresponded to non-glycosylated and glycosylated forms of EGFR-V5, respectively. The size of the latter band corresponded well with that of the EGFR expressed in mammalian cells. Indirect immunofluorescence analyses using confocal laser scanning microscopy (LSM) indicated that HA-EGF-FLO42 was successfully localized on the yeast cell surface (Figure 1d). After treatment with zymolyase, spheroplasts were also subjected to indirect immunofluorescence analyses that indicated that EGFR-V5 resided mainly in the yeast plasma membrane and partly in the cytoplasm (Figure 1e).

Expression and subcellular localization of the EGF-FLO42 and EGFR in yeast cells.
(a) Schematic drawing of the EGFR and FLO42-fused EGF in the periplasm of yeast cells. Upon binding EGF, the EGFR autophosphorylates. (b) A cell wall-anchored form of HA-tagged EGF (HA-EGF-FLO42) was expressed with the assistance of MFα1-prepro peptide. V5-tagged human EGFR (EGFR-V5) was expressed with the assistance of SUC2-signal peptide. TM, transmembrane domain. Numbers indicate amino acid residues at the borders of each domain. (c) Co-expression of HA-EGF-FLO42 and EGFR-V5 in yeast cells. Cytoplasmic (Cyto) and cell-wall (Wall) fractions were analyzed by western blotting using anti-HA (HA) and anti-V5 (V5) antibodies. Single and double asterisks indicate non-glycosylated and glycosylated forms, respectively. (d) Subcellular localization of HA-EGF-FLO42 in yeast cells. Intact cells were stained with anti-HA antibody. Bars = 2 μm. (e) Subcellular localization of EGFR-V5 in yeast cells. Spheroplasts were stained with anti-V5 antibody. Bars = 2 μm.
Localization of HA-EGF-FLO42 and EGFR-V5 on the yeast cell surface (seeFigure 1d and e) suggested that the EGFR was autophosphorylated by adjacent EGF in an autocrine manner. After incubation in HEPES buffer (pH 7.0) for 1 h to facilitate an efficient interaction between the EGFR and EGF, the fractions of cytoplasm and cell wall of yeast cells were prepared and then analyzed with anti-EGFR phosphotyrosine (pY) antibodies against pY-1068, pY-1148 and pY-1173 (Figure 2a). EGFR expressed in yeast cells was found to be phosphorylated by the co-expression of HA-EGF-FLO42 (lane 3), which was reduced by the treatment with tyrphostin AG147823, a specific inhibitor for EGFR autophosphorylation (lane 4). Next, yeast cells were treated with zymolyase and spheroplasts were probed with the anti-EGFR pY antibodies. Fluorescence derived from each phosphotyrosine was observed in cell peripheries (Figure 2c–e, left panels). Flow cytometric analyses confirmed that spheroplasts harboring phosphorylated EGFRs were substantially increased in an EGF-dependent manner (Figure 2c–e, right panels), whereas cell surface display of human interleukin-5 (HA-IL5-FLO42) (Figure 2b), a non EGFR-related protein, could not induce the phosphorylation of EGFR. Since anti-EGFR pY-1173 antibody showed good signal to noise ratio in western blot and flow cytometric analyses, we selected the antibody for further analyses. Taken together, cell surface engineering facilitated, for the first time, detection of EGF-dependent autophosphorylation of the EGFR in yeast cells, which allowed us to develop a new system to analyze ligand interactions with various receptors including RTKs and GPCRs without any interference from other mammalian proteins.

Autophosphorylation of EGFR-V5 induced by HA-EGF-FLO42 in yeast cells.
(a) EGF-dependent autophosphorylation of EGFR. Yeast cells co-expressing HA-EGF-FLO42 and EGFR-V5 were lysed and separated to cytoplasmic and cell wall fractions. Cytoplasmic fraction was subjected to western blot analysis using anti-V5 and anti-EGFR antibodies against phosphotyrosine-1068 (pY-1068), pY-1148 and pY-1173. Expression of HA-EGF-FLO42 in cell wall fraction was confirmed by using anti-HA antibody. Numbers shown at the bottom indicate lane number. Single and double asterisks indicate non-glycosylated and glycosylated forms, respectively. (b) Subcellular localization of HA-IL5-FLO42 in yeast cells. Intact cells were stained with anti-HA antibody. Bar = 2 μm. (c–e) Autophosphorylation of EGFR-V5 induced by HA-EGF-FLO42 in yeast cells. Spheroplasts were stained with a specific antibody against pY-1068 (c), pY-1148 (d) and pY-1173 (e) in the EGFR and then subjected to microscopic and flow cytometric analyses. HA-IL5-FLO42 was used as control of non-specific ligand for EGFR. Bars = 2 μm.
EGF-dependent homo-oligomerization of the EGFR in the yeast plasma membrane
As observed in mammalian cells24, autophosphorylation of the EGFR upon EGF-binding suggested homo-oligomer formation of EGFRs in the yeast plasma membrane. A split-ubiquitin (ub) membrane two-hybrid assay25 was carried out with the S. cerevisiae NMY51 strain harboring Lacz and HIS3 reporter genes under the control of the LexA-driven promoter using bait EGFR fused with the C-terminal half of ub (Cub) and a LexA-VP16 hybrid transcription factor (DNA-binding domain of LexA and transactivating domain of herpes simplex virus VP16) (EGFR-Cub-LexA-VP16) and prey EGFR fused with the N-terminal half of ub (Nub) (EGFR-Nub) (Supplementary Figure S1a). When ub was reconstituted upon oligomerization of the EGFR, the linkage between Cub and LexA was digested by endogenous ub C-terminal hydrolase, followed by release of LexA-VP16. By co-expression with HA-EGF-FLO42, yeast cells could grow on His-depleted plates and turn blue on X-gal-containing plates (Supplementary Figure S1b). These results indicated that EGF displayed on the yeast cell surface remarkably induced homo-oligomerization of EGFRs in the yeast plasma membrane.
Functional interaction of the EGFR with Grb2 and Shc1
By expressing constitutively active EGFR in yeast cells, autophosphorylated EGFRs recruited exogenous Grb2, Shc1 and Sos to the plasma membrane, followed by activation of the yeast RAS signaling pathway17. Therefore, we introduced HA-EGF-FLO42 and EGFR-V5 into a temperature-sensitive mutant of the S. cerevisiae cdc25h strain26 co-expressing the Sos-fused form of Grb2 or Shc1 (Supplementary Figure S2a). The cdc25h strain harbors temperature-sensitive CDC25 (CDC25ts) that functions as a GDP-GTP exchanging factor for RAS1 and RAS2 and induces growth inhibition at 37°C. When the EGFR was activated by EGF, yeast cells survived even at 37°C due to complementation of the function of CDC25ts by Grb2-Sos or Shc1-Sos (Supplementary Figure S2b). In control yeast cells expressing FLO42 instead of HA-EGF-FLO42, almost no cells could grow at 37°C. These results indicated that the yeast RAS signaling pathway was effectively activated by EGF-dependent EGFR autophosphorylation.
High-throughput de novo screening of EGFR agonists by an automated single-cell analysis and isolation system
Recently, we have developed an automated single-cell analysis and isolation system that isolates single cells of interest from arrays containing large number of cells19. Based on the functional interaction between EGF and the EGFR on the yeast cell surface, a conformationally constrained peptide library, instead of HA-EGF-FLO42, was introduced into yeast cells for de novo screening of EGFR agonists. The peptide library consisted of MFα1-prepro peptide, helix-loop-helix (HLH), FLAG-epitope tag and FLO42 from the N- to C-terminals (Figure 3a). The HLH moiety was composed of two helixes split by a hepta-glycine (G7)-loop, in which the C-terminal helix contained randomized sequences of five amino acids (approximately 3.2 × 106 variations) on its solvent-accessible surface17. Cells containing the peptide library were subjected to indirect immunofluorescence analysis with an anti-FLAG antibody and then observed by confocal LSM. Fluorescence was observed around the periphery of yeast cells (Figure 3b), similar to HA-EGF-FLO42 (seeFigure 1d), indicating that the peptide library was efficiently displayed on the yeast cell surface. Meanwhile, based on the flow cytometric analyses, about 100% of yeast cells expressed both HLH-FLAG-FLO42 library and EGFR-V5 concurrently, suggesting that randomized HLH peptide library could be screened thoroughly in yeast cell expressing EGFR-V5.

High-throughput de novo screening of EGFR agonists.
(a) A cell wall-anchored form of FLAG-tagged HLH (HLH-FLAG-FLO42) was expressed with the assistance of MFα1-prepro peptide. The HLH consisted of two α-helixes (blue cylinders) linked with a hepta-glycine (G7) loop, in which the second α-helix contained randomized sequences of five amino acids (red dots) on its solvent-accessible surface. (b) Subcellular localization of HLH-FLAG-FLO42 in yeast cells. Intact cells were stained with an anti-FLAG antibody. Bar = 2.5 μm. (c) Flowchart of EGFR agonist screening. Approximately 3.2 × 106 cells co-expressing HLH-FLAG-FLO42 (HLH) and EGFR-V5 (EGFR) were incubated at pH 7 for 1 h, fixed with paraformaldehyde and then treated with zymolyase, followed by incubation with an anti-phospho EGFR antibody. Approximately 2.5 × 105 cells were individually placed into the microchambers of cell array chip by brief centrifugation. The chip was scanned with a CCD camera under UV light by an automated single-cell analysis and isolation system. In the microchamber array chip, positive and negative cells were indicated by red and green circles, respectively. Positive cells were automatically retrieved with a glass capillary equipped on the micromanipulator. Using 20 chips, 83 positive cells were obtained and 65 DNA fragments were amplified by single cell-based PCR. Amino acid sequences were deduced from 13 fragments derived from cells with high fluorescence and then aligned. Finally, eight independent EGFR agonist candidates were obtained, synthesized in E. coli and then evaluated for autophosphorylation of the EGFR in A431 cells.
Yeast cells co-expressing the peptide library and EGFR were cultured for 16 h, incubated in HEPES buffer (pH 7.0) for 1 h, fixed with 4% PFA, treated with zymolyase, stained by indirect immunofluorescence with an anti-EGFR pY-1173 antibody and then placed into microchambers (a polystyrene slide containing 2.5 × 105 of 10-μm wells) by gentle centrifugation. About 80 ~ 90% of wells contained one to two spheroplasts. The fluorescent intensity and position of each spheroplast were analyzed by the automated single-cell analysis and isolation system (Figure 3c). After identifying positive spheroplasts with high fluorescent intensities, they were automatically retrieved with a glass capillary and individually transferred into the wells of a 96-well PCR plate. Finally, from approximately 5 × 106 spheroplasts (3.2 × 106 independent clones), 83 positive spheroplasts were isolated and their fluorescent intensity was already sorted in descending order.
Evaluation of EGFR agonist candidates in A431 cells
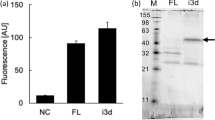
Using single cell-based PCR, 65 DNA fragments encoding the HLH moiety were successfully amplified from 83 positive spheroplasts (Figure 4a). Among 13 clones with high fluorescent intensities, DNA sequencing revealed that four (clones 2, 6, 9 and 10) and three clones (clones 8, 11 and 12) encoded identical amino acid sequences, respectively (Supplementary Tables S1 and S2). Eight clones encoding distinct amino acid sequences were expressed as an N-terminal thioredoxin-fused C-terminal hexahistidine-tagged form in Escherichia coli BL21 (DE3) (Figure 4b) and fused proteins were purified with a cobalt-chelating column (Figure 4c). These proteins were added to the culture medium of human adenosquamous carcinoma A431 cells that overexpress the EGFR. Western blot analysis using an anti-EGFR antibody against pY-1173 demonstrated that six proteins (derived from clones 2, 3, 5, 7, 8 and 13) possessed an agonistic activity for the EGFR in mammalian cells (Figure 5a), which was estimated less than 0.1% of EGF (working concentration, 1.5 nM for EGF versus 2.0 μM for peptides) (Figure 5b). However, the agonistic activity of each peptide was completely inhibited by the treatment with tyrphostin AG1478, corroborating that each peptide functions as an EGFR agonist.

Single-cell PCR cloning and bacterial expression of HLH proteins.
(a) Single-cell PCR. PCR products obtained by single-cell PCR were subjected to agarose gel electrophoresis. Numbers indicate clones amplified successfully (65 clones/83 lanes). M, DNA marker. Asterisks indicate the position of target bands. (b) Directional TOPO cloning. The cDNA fragments amplified by single cell PCR (216 bp) were extracted from agarose gel and then inserted into pET102 directional TOPO vector for the bacterial expression. Loop encodes hepta-glycine. Linker encodes N-terminal 20 amino acids of FLO42. Asterisks indicate the position of identical codon. (c) Purification of HLH proteins. HLH proteins expressed as an N-terminal thioredoxin-fused and C-terminal hexahistidine-tagged form in Escherichia coli BL21 (DE3) and purified with cobalt-chelating column. The purity was checked by CBB staining. Numbers indicate the clone number. M, protein marker. Asterisk indicates the position of target band.

Induction of EGFR autophosphorylation in A431 cells by EGFR agonist candidates.
(a) After treatment with 0.1 and 1.0 μM bacterially expressed HLH peptides (8 species, clones 2 ~ 17) for 5 and 20 min, A431 cell lysates were subjected to western blotting with anti-EGFR and anti-EGFR pY1173 antibodies. As a positive control, 1.5 nM EGF was used. (b) After 30-min treatment with 200 nM tyrphostin AG1478, A431 cells were treated with 2.0 μM HLH peptides for 20 min, lysed and subjected to western blotting with anti-EGFR and anti-EGFR pY-1173 antibodies. As a positive control, 1.5 nM EGF was used.
Discussion
Here, we demonstrated that the EGFR expressed in the yeast plasma membrane could form homo-oligomers upon stimulation with cell wall-anchored EGF, followed by its autophosphorylation and activation of the yeast RAS signaling pathway. This is the first report of yeast cells that can reconstitute a mammalian growth-signaling pathway mediated by the EGF-EGFR-Grb2/Shc1-Sos-Ras complex. By replacing EGF with a random peptide library, we achieved high-throughput screening of EGFR agonists using an automated single-cell analysis and isolation system to promptly identify six novel EGFR agonists that are active in A431 cells.
A cell wall is one of the major differences between yeast and mammalian cells. The thickness and pore size of the yeast cell wall are 50 ~ 200 nm27 and about 0.89 nm, respectively28. Because the size of the extracellular domain of the EGFR and the Stokes radius of EGF are about 11 nm29 and 3.2 nm30, respectively, extracellular proteinous ligands cannot reach the EGFR buried in the cell wall and ligands expressed in the vicinity of the yeast plasma membrane can interact with the EGFR. Thus, each yeast cell concurrently displaying the receptor and ligand candidates can work as ‘an independent and autonomous test tube’, allowing us to analyze a ligand-receptor interaction at a single cell level without exogenous interference. By displaying ligands on the yeast cell surface, molecules accumulate in the periplasm and cell wall31. Assuming that yeast cells are spheres of a 5.5 μm average diameter (n = 40) and the average thickness of the cell wall is 125 nm27, the volume of periplasmic and cell wall fractions is calculated as about 1.135 × 10−11 ml. Because the maximal number of small molecules displayed on the yeast cell surface is estimated to be about 106 per yeast cell32,33, the maximal concentration of cell wall-anchored EGF is estimated at 146 μM, which implies that a 1.2 × 105-fold of the EC50 of EGF (1.2 nM) to activate the EGFR in mammalian cells34. High concentrations of ligands on the yeast cell surface facilitate the identification of ligands, even if their agonistic activities are very low.
When yeast cells co-expressing the EGFR and HLH peptide library were established, we initially tried to isolate yeast cells with high fluorescence by fluorescence-activated cell sorting (FACS). Because conventional FACS systems only collect positive cells with a proportion of more than 0.1%19,35, we could not isolate positive clones (up to 100) from a peptide library of more than 1 × 106 by FACS. To address this issue, we used an automated single-cell analysis and isolation system that manipulates single cells arrayed on a chip with 250,000 microchambers. Consequently, 83 positive cells were isolated from about 3.2 × 106 independent clones, indicating that the automated single-cell analysis and isolation system can manipulate positive cells at a proportion of less than 0.0026%. In principle, the system can isolate yeast cells with the highest fluorescence from a peptide library of more than 106, which is nearly impossible by FACS.
A random peptide library with a flexible structure often weakly interacts with target molecules in an induced-fit manner36, leading to identification of false-positive molecules. Accordingly, we employed a conformationally constrained HLH peptide library for EGFR agonist screening. DNA sequence analyses revealed several independent clones encoding identical amino acid sequences (Supplementary Table S1) and about 85% of positive clones (11/13) could activate the EGFR in A431 cells, which strongly suggests that the HLH peptide library is reliable for drug screening. Previous studies with EGF mutants have revealed that some amino acid residues in EGF (Tyr-13, Leu-15, His-16, Arg-41, Gln-43 and Lue-47)37,38 together with Gly-1239 play a key role for EGFR binding and/or mitogenicity in mammalian cells (Supplementary Table S3). Furthermore, by using a fluorescence-labeled EGFR extracellular domain as a probe for EGF mutants displayed on the yeast cell surface, Cochran et al.40 and Lahti et al.41 have identified and analyzed various EGFR agonists, in which mutant 114, 28 and 123 possess over 30-fold affinity to EGFR compared with wild-type EGF40,41. When comparing the amino acid sequences among the previously reported EGF mutants and the HLH peptides harboring EGFR agonistic activity (seeSupplementary Table S3), the HLH peptides are fundamentally different from these EGF mutants not only in primary structure but also secondary structure. Thus, the biological function of each amino acid contained in the HLH and EGF mutants could not be completely compared. However, while some high affinity EGF mutants have been found to contain additional 1 ~ 4 Pro40, this amino acid residue was found in the identified HLH peptides at high frequency (4 of 6 agonistic HLH peptides; i.e., clone 2, 7, 8, 13). Since Pro is known to be α-helix breaker, the C-terminal α-helix of HLH peptides was presumably somewhat too straight to bind to EGFR structurally. On the other hand, HLH clone 4, which contains two Pro, did not show the EGFR agonistic activity, suggesting that the C-terminal α-helix of HLH peptides was essentially required to activate EGFR but should harbor a shorter or bent helix structure for more efficient EGFR activation. All EGFR agonist candidates selected by yeast-based screening were considered to possess high activity also in mammalian cells. Unexpectedly, two proteins (derived from clones 1 and 4) showed no agonistic activity and also 11 proteins showed low activity in A431 cells. This was probably caused by the differences of cellular conditions around EGFR between yeast cells and A431 cells and/or by insufficient contributions from randomized 5 amino acid residues located to C-terminal α-helix of the HLH peptides to bind to EGFR. If the screening of EGFR agonists with new peptide libraries (e.g., HLH scaffold harboring >6 randomized amino acid residues, new scaffold optimized for EGFR) is performed, new peptides with strong EGFR agonistic activity could be identified. Taken together, it is still worth performing the agonist screening in yeast cells for identifying lead compounds with novel structure in a high-throughput manner. Furthermore, as reported by Fujii et al.18, this library is suitable for extracting pharmacophores to design small chemical compounds by molecular dynamics simulation.
A single cell-based high-throughput drug screening system using yeast cell surface engineering to facilitate prompt de novo screening of lead compounds for rational drug design may accelerate the introduction of yeast technology to conventional mammalian cell-based drug discovery.
Methods
Plasmids
A DNA fragment encoding the yeast α-factor-prepro region (MFα1prepro, N-terminal 89 amino acids of the MFα1 precursor), N-terminal HA-tagged human EGF (HA-EGF) and yeast cell wall-anchored peptide FLO42 (C-terminal 42 amino acids of FLO1) was inserted under the control of the GAL1 promoter into pYES2 (URA3, Invitrogen, Carlsbad, CA, USA) (pYES2-MFα1prepro-HA-EGF-FLO42, seeFigure 1b). By replacing EGF-coding region with human IL5 gene, pYES2-MFα1prepro-HA-IL5-FLO42 was constructed for the expression of non EGFR-related ligand. A DNA fragment encoding the SUC2 signal peptide (25 amino acids), human EGFR (mature form, Leu-25 to Ala-1210 of the EGFR precursor, GenBank accession No. NM_005228.3) and C-terminal V5-tagged EGFR was inserted under the control of the GAL1 promoter into pYES3 (TRP1, Invitrogen) (pYES3-SUC-EGFR-V5, seeFigure 1b).
As a conformationally constrained peptide library, the DNA fragment encoding MFα1prepro, an intramolecular anti-parallel HLH peptide with a C-terminal FLAG tag and FLO42 was inserted under the control of the GAL1 promoter into pYES2 (pYES2-MFα1prepro-HLH-FLAG-FLO42). Briefly, primary PCR was carried out using 5′-CGC CAA GCT TGG GCG GAG CTC GCA GCT CTG GAA ATG GAA CTG GCG GCA CTC GAA GGT GGC GGC GGT GGT GGC GGC AAG CTG-3′ and 5′-T CCG CTC GAG TTT ATC ATC ATC ATC TTT ATA ATC GGA GCC TCC ACC AGC TTT CAA MNN MNN TAG CTT MNN TTT CAA MNN MNN CAG CTT GCC GCC ACC ACC-3′, in which N represents A, T, G or C and M represents A or C. The DNA fragment encoding randomized HLH-FLAG moiety was further amplified by using 5′-CGC CAA GCT TGG GCG GAG-3′ and 5′-TCC GCT CGA GTT TAT CAT CAT CAT CTT TAT-3′, treated with Hind III and Xho I and then inserted into pYES2-MFα1prepro-FLO42 vector. The HLH peptide contained randomized sequences of five amino acids on the solvent-accessible surface of the second helix.
To examine homo-oligomerization of the EGFR, the DNA fragment of the human EGFR (mature form) was inserted under the control of the ADH1 promoter of pPR3-SUC (TRP1, Mo Bi Tech, Göttingen, Germany) to express an N-terminal SUC2 signal peptide-fused and C-terminal Nub (N-terminal half of human ub)-fused form (pPR3-SUC-EGFR) and the same DNA fragment was inserted under the control of the CYC1 promoter into pBT3-SUC (LEU2, Mo Bi Tech) to express an N-terminal SUC2 signal peptide-fused and C-terminal Cub (C-terminal half of human ub)-LexA (DNA-binding domain)-VP16 (transactivating domain)-fused form (pBT3-SUC-EGFR).
To examine interactions of the EGFR with Grb2 and Shc1, the DNA fragment encoding MFα1prepro and N-terminal FLAG-tagged EGFR was inserted under the control of the GAL1 promoter of pGMH20 (HIS3, RIKEN Bioresource Center, Ibaraki, Japan) (pGMH20-MFα1prepro-FLAG-EGFR). Human Grb2 cDNA (NM_002086.4) and human Shc1 cDNA (NM_183001.3) were inserted under the control of the ADH1 promoter of the pSos plasmid (Stratagene, La Jolla, CA, USA) to express an N-terminal Sos (N-terminal 1063 amino acids of Sos)-fused form of Grb2 (pSos-Grb2) and Shc1 (pSos-Shc1), respectively.
Yeast strains
S. cerevisiae BJ5464 (MATα ura3-52 his3-200 trp1 leu2-1 pep4::HIS3 prb1Δ1.6R can1 Gal+) was obtained from the Yeast Genetic Resource Center (Osaka, Japan). S. cerevisiae cdc25h (MATa ura3-52 his3-200 trp1-901 leu2-3,112 ade2-101 lys2-801 cdc25-2 Gal+) and NMY51 (MATa his3-200 trp1-901 leu2-3,112 ade2 ura3::(lexAop)8-lacZ LYS2::(lexAop)4-HIS3 (lexAop)8-ADE2 GAL4) were purchased from Stratagene and Mo Bi Tech, respectively. Manipulation of yeast cells was performed according to the yeast protocol handbook (Clontech).
Western blotting
Yeast cells were incubated with SD medium containing 20 mM HEPES (pH 7.0) for 1 h to allow interaction between EGF and the EGFR on the yeast cell surface. When using tyrphostin AG1478 (N-(3-chlorophenyl)-6,7-dimethoxy-4-quinazolinamine) (Cell Signaling Technology, Beverly, MA, USA), cells were treated with the reagent at 1 μM for 30 min before lysis. Yeast cells were suspended in a lysis buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1 mM EGTA, 1% (v/v) Triton X-100, 2 mM dithiothreitol and 1 tablet per 10 ml Complete™ protease inhibitor cocktail (Roche, Basel, Switzerland), frozen in liquid nitrogen and then disrupted with vigorous shaking in an automatic homogenizer (Automill™; Tokken, Chiba, Japan). After centrifugation, the supernatant was collected as the cytosolic fraction. The cell-wall fraction was extracted from cell debris with 0.5% (w/v) SDS and 1% (v/v) 2-mercaptomethanol42. Each fraction was subjected to SDS-PAGE followed by western blotting with a horseradish peroxidase (HRP)-conjugated anti-HA antibody (clone 12CA5, Roche), an HRP-conjugated anti-V5 antibody (clone V5–10, Sigma-Aldrich, St Louis, MO, USA), anti-EGFR pY-1068 mouse monoclonal antibody (clone 15A2, nanoTools, Teningen, Germany), anti-EGFR pY-1148 rabbit polyclonal antibody (Novus Biologicals, Littleton, CO, USA), anti-EGFR pY-1173 goat polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and appropriate HRP-conjugated secondary antibodies (anti-mouse IgG (GE Healthcare, St. Giles), anti-rabbit IgG (GE Healthcare), anti-goat IgG (Santa Cruz)). Immunoreactive bands were visualized with an ECL Plus Western Blotting Detection System (GE Healthcare).
Immunocytochemical analysis
Yeast cells were incubated with SD medium containing 20 mM HEPES (pH 7.0) for 1 h to allow interaction between EGF and the EGFR on the yeast cell surface. Cells were incubated in fixation buffer (40 mM potassium phosphate, pH 7.2, 4% (w/v) paraformaldehyde (PFA) at room temperature for 30 min, treated with 12 U/ml zymolyase 100 T (Seikagaku, Tokyo, Japan) in sorbitol buffer (40 mM potassium phosphate, pH 7.2, 1.2 M sorbitol and 0.5 mM MgCl2) to produce spheroplasts, incubated in blocking buffer (10 mM Tris-HCl, pH 7.2, 150 mM NaCl and 2% (w/v) bovine serum albumin (BSA) and then labeled with appropriate primary antibodies at room temperature for 1 h. To detect HA-EGF-FLO42 and HA-IL5-FLO42, intact cells were used instead of spheroplasts. Labeled cells were washed with TBS (10 mM Tris-HCl, pH 7.2 and 150 mM NaCl), incubated in blocking buffer containing Alexa Fluor 488-labeled anti-IgG (Invitrogen) at room temperature for 30 min, washed with TBS and then analyzed with a FLUOVIEW FV1000 confocal laser scanning microscope (Olympus, Tokyo, Japan) and FACSCanto™ II flow cytometer (BD Biosciences, San Jose, CA, USA). Antibodies used were as follows: anti-HA (clone 12CA5), anti-V5 (clone V5005, Nacalai, Kyoto, Japan), anti-EGFR phosphotyrosine-1068 (pY-1068) (clone 15A2, nanoTools, Teningen, Germany), anti-EGFR pY-1148 (Novus Biologicals, Littleton, CO, USA) and anti-EGFR pY-1173 (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Split-ub membrane two-hybrid assay
S. cerevisiae NMY51 harboring pPR3-SUC-EGFR (pray), pBT3-SUC-EGFR (bait) and either pYES2-MFα1prepro-HA-EGF-FLO42 (ligand) or pYES2-MFα1prepro-FLO42 (control) were plated on a His-depleted SD plate or an X-Gal-containing SD plate and incubated at 30°C for 8 days.
Complementation assay with the CDC25ts strain
S. cerevisiae cdc25h harboring pGMH20-MFα1prepro-FLAG-EGFR, either pSos-Grb2 or pSos-Shc1 and either pYES2-MFα1prepro-HA-EGF-FLO42 (ligand) or pYES2-MFα1prepro-FLO42 (control) were plated on an SD plate and incubated at 25 or 37°C for 7 days.
De novo agonist screening with an automated single-cell analysis and isolation system
S. cerevisiae BJ5464 cells harboring pYES2-MFα1prepro-HLH-FLAG-FLO42 and pYES3-SUC-EGFR-V5 were incubated with SD medium containing 20 mM HEPES (pH 7.0) for 1 h, fixed with PFA, treated with zymolyase, blocked with BSA and then stained with an anti-EGFR pY1173 antibody. Approximately 2.5 × 105 fixed spheroplasts in 1 ml PBS were individually placed into the microchambers (10 μm diameter, 30 μm well-to-well pitch and 10 μm well depth) of a cell array chip (256,000 wells; 20 × 20 wells/subarea; 20 × 32 subareas/chip on a 1.39 × 2.23 cm2 chip) by brief centrifugation (7 × g at room temperature for 1 min, three times) and then analyzed with the automated single-cell analysis and isolation system (As One corp., Osaka, Japan). Each cell with high fluorescence was collected by the automated single-cell analysis and isolation system in descending order of fluorescent intensity as described previously19. Briefly, microchambers containing no or more than 2 fluorescent spheroplasts were excluded from further analyses. A histogram together with a list of correlations between the position and fluorescent intensity of each spheroplast was generated. Target spheroplasts could be virtually marked in descending order of fluorescent intensity. Spheroplasts of interest were automatically collected with a glass capillary attached to the micromanipulator of the automated single-cell analysis and isolation system, which were optimized not to pick up the adjacent non-target spheroplasts and confirmed by elimination of fluorescence in the target microchamber automatically. Upon failure, the system automatically repeated the collection process. Each spheroplast was transferred and released into the assigned well in 96-well PCR plates. The reciprocal movement of the glass capillary required 15 s for each spheroplast. After repeating the procedure 20 times, 83 spheroplasts as positive clones isolated from about 3.2 × 106 independent clones were moved to a 96 well PCR plate and then subjected to single cell-based PCR. Primary PCR was carried out using 5′-GCC TTA TTT CTG GGG TAA TTA ATC AGC G-3′, 5′-ACC TAG ACT TCA GGT TGT CTA ACT CCT TCC-3′ and KOD-Plus- (TOYOBO, Osaka, Japan) to amplify the DNA fragment encoding the GAL1 promoter, MFα1prepro-HLH-FLAG-FLO42 and CYC1 terminator. The thermal cycling of the primary PCR was pre-heating at 94°C for 5 min; 12 cycles of denaturing at 94°C for 1 min; annealing from 73°C to 61°C (–1°C/cycle) for 20 sec; extension at 68°C for 90 sec; followed by 45 cycles of denaturing at 94°C for 1 min; annealing at 64°C for 30 sec; extension at 68°C for 90 sec and final extension at 68°C for 7 min. The secondary PCR was carried out using 5′-CAC CCA AGC TTG GGC GGA GCT CGC AGC TCT-3′, 5′-GGC ACT GCC AGC ATA CGT TGA AAT-3′ and KOD-Plus- to amplify the DNA fragment encoding HLH-FLAG and the N-terminal 20 amino acids of FLO42. The thermal cycling of the secondary PCR was pre-heating at 94°C for 2 min; 12 cycles of denaturing at 94°C for 1 min; annealing from 77°C to 63°C (–1°C/cycle) for 20 sec; extension at 68°C for 15 sec; followed by 45 cycles of denaturing at 94°C for 1 min; annealing at 66°C for 30 sec; extension at 68°C for 15 sec and final extension at 68°C for 7 min. The amplified DNA fragments were inserted under the control of the T7 promoter in the pET102/D-TOPO vector (Invitrogen) (pET102-HLH-FLAG).
Bacterial expression of EGFR agonist candidates
E. coli BL21 (DE3) harboring pET102-HLH-FLAG was cultured in an Overnight Express Autoinduction System (Merck, Darmstadt, Germany). N-terminal thioredoxin (TRX)-fused and C-terminal His6-tagged forms of HLH-FLAG were purified by the BD TALON™ HT 96-Well Purification System (BD) according to the manufacture's protocol. Homogeneity of TRX-HLH-FLAG-His6 protein was confirmed by SDS-PAGE followed by Coomassie brilliant blue R-250 staining.
Autophosphorylation assay in A431 cells
Human squamous carcinoma A431 cells (approximately 5 × 104 cells) cultured on a 3.5 cm dish were incubated in serum-free medium for 16 h and then treated with 0.1, 1.0, or 2.0 μM TRX-HLH-FLAG-His6 proteins for 5 and 20 min. As a positive control, 1.5 nM recombinant human EGF (PEPROTECH, Rocky Hill, NJ, USA) was used. When using tyrphostin AG1478, cells were treated with the reagent at 200 nM for 30 min before ligand (i.e., EGF, HLH) treatment. Cells were lysed with lysis buffer (50 mM Tris-HCl, pH 7.2, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, 1 mM Na3VO4, 1% (v/v) phosphatase inhibitor cocktail I (SIGMA) and 1 tablet/50 ml Complete™ (Roche), mixed with 1 × Laemmli's sample buffer and then boiled for 15 min. Samples were analyzed by western blotting using anti-EGFR (Rockland, Gilbertsville, PA, USA) and anti-EGFR pY-1173 antibodies.
References
Smith, C. & Teitler, M. Beta-blocker selectivity at cloned human beta 1- and beta 2-adrenergic receptors. Cardiovasc. Drugs Ther. 13, 123–126 (1999).
Lazou, A., Sugden, P. H. & Clerk, A. Activation of mitogen-activated protein kinases (p38-MAPKs, SAPKs/JNKs and ERKs) by the G-protein-coupled receptor agonist phenylephrine in the perfused rat heart. Biochem. J. 332, 459–465 (1998).
Sirotnak, F. M., Zakowski, M. F., Miller, V. A., Scher, H. I. & Kris, M. G. Efficacy of cytotoxic agents against human tumor xenografts is markedly enhanced by coadministration of ZD1839 (Iressa), an inhibitor of EGFR tyrosine kinase. Clin. Cancer Res. 6, 4885–4892 (2000).
Slamon, D. J. et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 344, 783–792 (2001).
Costa-Pereira, A. P. et al. Mutational switch of an IL-6 response to an interferon-gamma-like response. Proc. Natl. Acad. Sci. USA 99, 8043–8047 (2002).
Velloso, L. A. et al. Cross-talk between the insulin and angiotensin signaling systems. Proc. Natl. Acad. Sci. USA 93, 12490–12495 (1996).
Suzuki, A. et al. CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 193, 471–481 (2001).
Antonio, C. N., Grazia, P. M., Marialessandra, C., Francesco, B. & Roberto, P. Receptor-drug interaction: europium employment for studying the biochemical pathway of g-protein-coupled receptor activation. Met. Based Drugs 2007, 12635 (2007).
Luque, R. M. et al. Differential contribution of nitric oxide and cGMP to the stimulatory effects of growth hormone-releasing hormone and low-concentration somatostatin on growth hormone release from somatotrophs. J. Neuroendocrinol. 17, 577–582 (2005).
Finch, E. A., Turner, T. J. & Goldin, S. M. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science 252, 443–446 (1991).
Naider, F. & Becker, J. M. The alpha-factor mating pheromone of Saccharomyces cerevisiae: a model for studying the interaction of peptide hormones and G protein-coupled receptors. Peptides 25, 1441–1463 (2004).
Minic, J., Sautel, M., Salesse, R. & Pajot-Augy, E. Yeast system as a screening tool for pharmacological assessment of g protein coupled receptors. Curr. Med. Chem. 12, 961–969 (2005).
Miret, J. J., Rakhilina, L., Silverman, L. & Oehlen, B. Functional expression of heteromeric calcitonin gene-related peptide and adrenomedullin receptors in yeast. J. Biol. Chem. 277, 6881–6887 (2002).
Floyd, D. H. et al. C5a receptor oligomerization. II. Fluorescence resonance energy transfer studies of a human G protein-coupled receptor expressed in yeast. J. Biol. Chem. 278, 35354–35361 (2003).
Klein, C. et al. Identification of surrogate agonists for the human FPRL-1 receptor by autocrine selection in yeast. Nat. Biotechnol. 16, 1334–1337 (1998).
Manning, G., Plowman, G. D., Hunter, T. & Sudarsanam, S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 27, 514–520 (2002).
Busti, S., Sacco, E., Martegani, E. & Vanoni, M. Functional coupling of the mammalian EGF receptor to the Ras/cAMP pathway in the yeast Saccharomyces cerevisiae. Curr. Genet. 53, 153–162 (2008).
El-Haggar, R. et al. Molecular design of small organic molecules based on structural information for a conformationally constrained peptide that binds to G-CSF receptor. Bioorg. Med. Chem. Lett. 20, 1169–1172 (2010).
Yoshimoto, N. et al. An automated system for high-throughput single cell-based breeding. Sci. Rep. 3, 1191 (2013).
Emr, S. D., Schekman, R., Flessel, M. C. & Thorner, J. An MF alpha 1-SUC2 (alpha-factor-invertase) gene fusion for study of protein localization and gene expression in yeast. Proc. Natl. Acad. Sci. USA 80, 7080–7084 (1983).
Sato, N. et al. Long anchor using Flo1 protein enhances reactivity of cell surface-displayed glucoamylase to polymer substrates. Appl. Microbiol. Biotechnol. 60, 469–474 (2002).
Bielefeld, M. & Hollenberg, C. P. Bacterial beta-lactamase is efficiently secreted in Saccharomyces cerevisiae under control of the invertase signal sequence. Curr. Genet. 21, 265–268 (1992).
Johns, T. G. et al. Antitumor efficacy of cytotoxic drugs and the monoclonal antibody 806 is enhanced by the EGF receptor inhibitor AG1478. Proc. Natl. Acad. Sci. USA 100, 15871–15876 (2003).
Ferguson, K. M. et al. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 (2003).
Stagljar, I., Korostensky, C., Johnsson, N. & te Heesen, S. A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo. Proc. Natl. Acad. Sci. USA 95, 5187–5192 (1998).
Petitjean, A., Hilger, F. & Tatchell, K. Comparison of thermosensitive alleles of the CDC25 gene involved in the cAMP metabolism of Saccharomyces cerevisiae. Genetics 124, 797–806 (1990).
Linnemans, W. A., Boer, P. & Elbers, P. F. Localization of acid phosphatase in Saccharomyces cerevisiae: a clue to cell wall formation. J. Bacteriol. 131, 638–644 (1977).
De Nobel, J. G. et al. Cyclic variations in the permeability of the cell wall of Saccharomyces cerevisiae. Yeast 7, 589–598 (1991).
Lemmon, M. A. et al. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 16, 281–294 (1997).
Nexø, E., Jørgensen, P. E., Thim, L. & Roepstorff, P. Purification and characterization of a low and a high molecular weight form of epidermal growth factor from rat urine. Biochim. Biophys. Acta 1037, 388–393 (1990).
Jin, Y. L. & Speers, R. A. Flocculation of Saccharomyces cerevisiae. Food Res. Int. 31, 421–440 (1998).
Ueda, M. & Tanaka, A. Genetic immobilization of proteins on the yeast cell surface. Biotechnol. Adv. 18, 121–140 (2000).
Takayama, K. et al. Surface display of organophosphorus hydrolase on Saccharomyces cerevisiae. Biotechnol. Prog. 22, 939–943 (2006).
Gilmore, J. L., Gallo, R. M. & Riese, D. J., 2nd The epidermal growth factor receptor (EGFR)-S442F mutant displays increased affinity for neuregulin-2beta and agonist-independent coupling with downstream signalling events. Biochem. J. 396, 79–88 (2006).
Yamamura, S. et al. Single-cell microarray for analyzing cellular response. Anal. Chem. 77, 8050–8056 (2005).
Rini, J. M., Schulze-Gahmen, U. & Wilson, I. A. Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science 255, 959–965 (1992).
Campbell, I. D., Cooke, R. M., Baron, M., Harvey, T. S. & Tappin, M. J. The solution structures of epidermal growth factor and transforming growth factor alpha. Prog. Growth Factor Res. 1, 13–22 (1989).
Souriau, C., Gracy, J., Chiche, L. & Weill, M. Direct selection of EGF mutants displayed on filamentous phage using cells overexpressing EGF receptor. Biol. Chem. 380, 451–458 (1999).
Mullenbach, G. T. et al. Modification of a receptor-binding surface of epidermal growth factor (EGF): analogs with enhanced receptor affinity at low pH or at neutrality. Protein Eng. 11, 473–480 (1998).
Cochran, J. R., Kim, Y. S., Lippow, S. M., Rao, B. & Wittrup, K. D. Improved mutants from directed evolution are biased to orthologous substitutions. Protein Eng. Des. Sel. 19, 245–253 (2006).
Lahti, J. L. et al. Engineered epidermal growth factor mutants with faster binding on-rates correlate with enhanced receptor activation. FEBS Lett. 585, 1135–1159 (2011).
Schreuder, M. P., Brekelmans, S., van den Ende, H. & Klis, F. M. Targeting of a heterologous protein to the cell wall of Saccharomyces cerevisiae. Yeast 9, 399–409 (1993).
Acknowledgements
We are grateful to Prof. Hidekatsu Iha (Oita University) for pGMH20 and pGMT20 vectors. This study was supported in part by Adaptable & Seamless Technology Transfer Program through Target-driven R&D (A-STEP) by the Japan Science and Technology Agency (JST) (AS231Z04687F to N.Y., AS2311699F to S.K.), the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (H22-7, BRAIN to S.K.), a Grant-in-Aid for Scientific Research (A) (25242043 to S.K.), the Health Labor Sciences Research Grant from the Ministry of Health Labor and Welfare (to S.K.) and the As One Corporation (to S.K.). We thank Jun Ishii (Kobe University), Ken-ichi Kimura and Xu Jie (Furukawa), Masato Fujihashi and Masaya Kurokawa (STARLITE) and Masahiro Matsushita, Kenji Uemukai and Tohru Kaneno (As One).
Author information
Authors and Affiliations
Contributions
N.Y., K.Tatematsu, I.F., A.K., K.Tanizawa and S.K. designed research; N.Y., K.Tatematsu, M.I., T.N. and A.D.M. performed research; N.Y., K.Tatematsu and S.K. analyzed data; and N.Y. and S.K. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Yoshimoto, N., Tatematsu, K., Iijima, M. et al. High-throughput de novo screening of receptor agonists with an automated single-cell analysis and isolation system. Sci Rep 4, 4242 (2014). https://doi.org/10.1038/srep04242
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep04242
This article is cited by
-
Hierarchical design of artificial proteins and complexes toward synthetic structural biology
Biophysical Reviews (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
