
Abstract
Many risk factors/interventions in epidemiologic/biomedical studies are of minuscule effects. To detect such weak associations, one needs a study with a very large sample size (the number of subjects, n). The n of a study can be increased but unfortunately only to an extent. Here, we propose a novel method which hinges on increasing sample size in a different direction–the total number of variables (p). We construct a p-based ‘multiple perturbation test’ and conduct power calculations and computer simulations to show that it can achieve a very high power to detect weak associations when p can be made very large. As a demonstration, we apply the method to analyze a genome-wide association study on age-related macular degeneration and identify two novel genetic variants that are significantly associated with the disease. The p-based method may set a stage for a new paradigm of statistical tests.
Similar content being viewed by others

Introduction
Many risk factors/interventions in epidemiologic/biomedical studies are of minuscule effects1. For example, television viewing was found to increase the risks of type 2 diabetes, cardiovascular disease and all-cause mortality, but the effects in terms of relative risks are small: 1.20, 1.15 and 1.132, respectively; regular supplement of vitamin C was associated with a shortening of the duration of common colds, but with a relative risk (0.92) very near unity3. Moving into this ‘–omics’ era, for the first time researchers are becoming able to probe into study subjects' genome, transcriptome and metabolome, etc, to search for possible disease associations. However, the associations found so far were still very weak; for example the great majority of the odds ratios of genetic polymorphisms in genome-wide association studies were less than 1.54,5.
To detect weak associations, a very large sample size is needed. For example, in genome-wide association studies, the sample sizes have steeply increased from a few hundreds in the first study of age-related macular degeneration6 to tens of thousands in recent meta-analyses7,8. Also, the consortium-based studies are becoming increasingly indispensible as the single-institution studies often cannot meet the tough sample-size requirements. For example, the Wellcome Trust Case-Control Consortium9, the United Kingdom Biobank10 and China Kadoorie Biobank11 have recruited study subjects in the order of hundreds of thousands. But how big is big enough for sample size? A simulation study suggested that in some scenarios the sample size needed can easily go up to the millions!12 Certainly, there is a limit for the total number of subjects any research institution, any meta-analysis and any consortium can possibly assemble.
Traditionally, sample sizes are measured in terms of the total number of study subjects (n). In this study, we propose a novel ‘p-based’ method which hinges on increasing sample size in a different direction–the total number of variables (p).We construct a p-based ‘multiple perturbation test’ and conduct theoretical power calculations and computer simulations to show that it can achieve a very high power to detect a weak association when p can be made very large, say, to the thousands, millions or even more. We will also apply the new method to re-analyze a published genome-wide association study.
Results
Sharp null
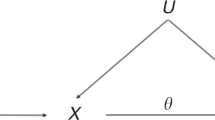
Assume that we are interested in the association between a binary factor, X (X = 1: exposed; X = 0: unexposed) and a disease, D (D = 1: diseased; D = 0: non-diseased). Consider also a binary auxiliary variable, Z (Z = 1 or 0), which is not of direct interest to us, but may help discern the possible association between X and D. Our method is based on testing whether the disease risk varies with X in any segment of the population demarcated by Z, i.e., testing the ‘sharp null’,

for both Z = 1 and Z = 0, against the alternative,

for either Z = 1 or Z = 0.
In a case-control study conducted in the study population, the Online Methods section shows that testing the sharp null amounts to testing the equality of odds ratios of X and Z, between the case group ( ) and the control group (
) and the control group ( ), or equivalently, testing whether there is an ‘interaction’ between X and Z with regard to the risk of D on a multiplicative scale:
), or equivalently, testing whether there is an ‘interaction’ between X and Z with regard to the risk of D on a multiplicative scale:

The following test statistic is proposed (see Supplementary Table S1 for the cell counts):

where j and k indicate the statuses of X and Z, respectively and  and
and  denote the numbers of case and control subjects with (X = j, Z = k), respectively.
denote the numbers of case and control subjects with (X = j, Z = k), respectively.  is distributed asymptotically as a df = 1 chi-squared distribution under the sharp null.
is distributed asymptotically as a df = 1 chi-squared distribution under the sharp null.
Essentially,  is testing whether the observed
is testing whether the observed  and
and  are being ‘perturbed’ too much away from
are being ‘perturbed’ too much away from  (the population odds ratio of X and Z and the expected value for both
(the population odds ratio of X and Z and the expected value for both  and
and  under the sharp null) than chance alone would dictate. We therefore refer to it as a ‘perturbation test’.
under the sharp null) than chance alone would dictate. We therefore refer to it as a ‘perturbation test’.
Multiple perturbation test
One single auxiliary variable may not perturb the above odds ratios very much. But if one has a whole panel of auxiliary variables (the Zi and the corresponding  , for i = 1, 2, …, p), one can construct a very powerful multiple perturbation test (MPT), by summing up the perturbations from the many auxiliary variables (Zs) in the panel:
, for i = 1, 2, …, p), one can construct a very powerful multiple perturbation test (MPT), by summing up the perturbations from the many auxiliary variables (Zs) in the panel:

MPT as such is a p-based test. Its power to detect a non-null X should increase as more Zs are included in the panel (as p increases). On the other hand, a truly innocent X should be able to stand the test from multiple Zs, even if p goes to infinity.
Figure 1 compares the theoretical powers of MPT and  (the conventional n-based test for the ‘crude null’). For
(the conventional n-based test for the ‘crude null’). For  , we need a very large study (n = ~15,000) to attain an adequate power of 80%. On the other hand, the power of MPT increases with p, surpasses that of
, we need a very large study (n = ~15,000) to attain an adequate power of 80%. On the other hand, the power of MPT increases with p, surpasses that of  and then can reach ~100% if p is sufficiently large. Supplementary Figure S1 shows that to make up for the power loss in using dependent Zs, one can simply include more Zs in the panel. Supplementary Table S2 shows that MPT can maintain accurate type I error rates for all scenarios considered.
and then can reach ~100% if p is sufficiently large. Supplementary Figure S1 shows that to make up for the power loss in using dependent Zs, one can simply include more Zs in the panel. Supplementary Table S2 shows that MPT can maintain accurate type I error rates for all scenarios considered.

Powers of MPT for the sharp null (solid lines, theoretical power assuming independent auxiliary variables with perturbation proportion of, from left to right respectively, π = 1.0, 0.2, 0.1 and 0.05) and the conventional test for the crude null (dashed line), under different number of subjects (a: n = 500, b: n = 1,000, c: n = 5,000) and number of auxiliary variables. The power of the n-based  increases with n. The power gain is only 30%, from 8% (n = 500, a) to 38% (n = 5,000, c). The power of the p-based MPT increases with p in all scenarios that we considered and surpasses the power of
increases with n. The power gain is only 30%, from 8% (n = 500, a) to 38% (n = 5,000, c). The power of the p-based MPT increases with p in all scenarios that we considered and surpasses the power of  when p ≈ 3,000 for π = 1, p ≈ 60,000 for π = 0.2, p ≈ 250,000 for π = 0.1 and p ≈ 1,000,000 for π = 0.05. Under π = 1, the power of MPT can reach nearly 100% when p is sufficiently large (p > ~1,000,000 when n = 500; p > ~100,000 when n = 1,000; p > ~10,000 when n = 5,000). Under π < 1, ~100% power is also possible if p can be made even larger.
when p ≈ 3,000 for π = 1, p ≈ 60,000 for π = 0.2, p ≈ 250,000 for π = 0.1 and p ≈ 1,000,000 for π = 0.05. Under π = 1, the power of MPT can reach nearly 100% when p is sufficiently large (p > ~1,000,000 when n = 500; p > ~100,000 when n = 1,000; p > ~10,000 when n = 5,000). Under π < 1, ~100% power is also possible if p can be made even larger.
The proposed MPT is applied to a public-domain data from a genome-wide association study of age-related macular degeneration6. Based on the data of chromosome 1 [a total 6639 single nucleotide polymorphisms (SNPs); p (the number of auxiliary variables) = 6638 for each SNP], the method detects two significant SNPs at false discovery rate (FDR)13 of 0.05: rs2618034 (q-value = 0.026) and rs2014029 (q-value = 0.045) (Table 1). These two SNPs clearly stand out in the Manhattan plot (Supplementary Fig. S2). We deliberately reduce the number of auxiliary variables (p = 3000, randomly selected from 6639 SNPs). The two SNPs remain at the top, though not reaching significance (Supplementary Fig. S3). On the other hand, we expand the number of auxiliary variables (p > 6638, randomly selected from chromosome 2 to chromosome 22). The two SNPs are still significant (Supplementary Table S3).
Figure 2 shows the fixation and drifting of P-values of the MPT. Although the 3rd top SNP (rs437749) is not significant by our FDR standard (Table 1), it is already displaying a fixation pattern in our fixation/drifting analysis (Fig. 2c). This suggests that if we can incorporate more perturbation SNPs into the MPT, SNP rs437749 may become significant. We deliberately remove the respective five largest  's in the MPTs for the two significant SNPs. Even so, a clear fixation pattern can still be seen for both (Supplementary Fig. S4).
's in the MPTs for the two significant SNPs. Even so, a clear fixation pattern can still be seen for both (Supplementary Fig. S4).

Fixation ((a–c), respectively for the 1st to the 3rd top SNPs on chromosome 1) and drifting ((d–f), for three purposefully chosen middle-to-bottom ranking SNPs on chromosome 1) of the P-values of MPT when only a certain number of perturbation SNPs are randomly incorporated for the age-related macular degeneration data. Each panel includes three lines (solid, dashed and dotted) representing three random incorporation sequences.
Each P-value is obtained from 1,000,000 rounds of permutation. The P-values initially fluctuate a lot, when the number of perturbation SNPs incorporated is small. But beyond a certain point, the P-values become ‘fixed’ exactly to the abscissa (P-values = 0) (a and b), or almost so (P-values ≈ 0) (c). By comparison, the P-values of all three purposefully chosen middle-to-bottom ranking SNPs are ‘drifting’ all the way without showing any sign of a fixation (d–f).
We also test run the proposed MPT on chromosome 19 (see Supplementary Note). Again, MPT proves to be very powerful. With FDR controlled at 0.05, it detects two significant SNPs (rs862703 and rs302437) (Supplementary Table S4) which also show fixations of P-values (Supplementary Fig. S5) and significantly stand out in the Manhattan plot (Supplementary Fig. S6).
Discussion
While confronted with high-throughput data, researchers often turn to dimension reduction methods to ease the severe penalty associated with testing myriads of variables14,15,16,17,18. For our p-based method, dimensionality is not a curse but in fact is a blessing. We see that the power of the MPT actually increases as the number of auxiliary variables increases. Such ‘the-more-the-better’ principle also applies, when one is knowledgeable about which variables may be perturbative. In Figure 3, since the initial power is only 0.59, should researchers add more variables into the test? We see as expected that adding more variables unselectively into the test will only dilute the power. However, upon more and more of low-informativity variables being added, the power can rise up again and then surpasses the original power.

Power curve when a researcher includes the 100 informative variables (I = 0.02) known to him/her and then other low-informativity variables (dotted lines from left to right, for I = 0.001, 0.00025 and 0.0001, respectively) unselectively into MPT.
However, the p-based approach only goes so far as when the auxiliary variables have a non-zero informativeness (I > 0, irrespectively of how small it may be). A computer can easily generate millions and billions of random variables for us, but all these artificial data amount to nothing (I = 0, exactly). The more such variables being added, the more the power will be curtailed. Another caveat is that there is no use replicate the data at hand just to make the total number of auxiliary variables appear larger; the power simply won't budge with this maneuver.
Age-related macular degeneration is a progressive disease in macula of the retina in which the pigment epithelium cells and the photoreceptor cells degenerate, causing gradual loss of central vision19,20. With FDR controlled at 0.05, in this study we are able to identify two novel SNPs on chromosome 1 that are significantly associated with the disease. The first SNP (rs2618034) is located in the intron region of KCND3 gene (potassium voltage-gated channel, Shal-related subfamily, member 3) on chromosome 1p13.2 and the second (rs2014029), the intron region of DTL gene (denticleless E3 ubiquitin protein ligase homolog (Drosophila)) on 1q32.3. KCND3 gene encodes Kv4.3 regulating neuronal excitability21. Mutations in KCND3 gene have been identified as a cause for cerebellar neurodegeneration22,23. In this regard, it is worthy to note that the retina photoreceptor cells are a specialized type of neurons which may also degenerate with aging. Meanwhile, DTL gene regulates p53 polyubiquitination and protein stability24 and the evidence to date suggests that p53 is a key regulator involved in the apotosis of retinal pigment epithelium cells25. All these findings further support that KCND3 and DTL genes may be causally related to the development of age-related macular degeneration. [As regards the two significant SNPs found on chromosome 19, their associations with age-related macular degeneration are also biologically plausible (see Supplementary Note)].
The multiple perturbation test indeed is a very powerful test. The two significant SNPs on chromosome 1 (rs2618034 and rs2014029) that we identified in this study are only very weakly associated with age-related macular degeneration (marginal association odds ratios = 0.53 and 2.10, respectively) and the traditional n-based method (Pearson chi-square test) comes nowhere near detecting them (P-values = 0.201 and 0.166, respectively) (Table 1). Even if we increase the total number of subjects from the present n = 146 (Klein et al.'s data6) to n ≈ 25,000 and n ≈ 77,000 (Holliday et al.'s7 and Fritsche et al.'s8 meta-analyses data), the n-based method still cannot detect them. But this is not to say that the n-based method is useless. In fact, Klein et al.6 themselves presented one SNP (rs380390) with an n-based P-value of 4.1 × 10−8 (significance after Bonferroni correction), but it is undetectable with our method. The p-based MPT is good at detecting interactive associations, i.e., associations that are prone to be perturbed by other factors, regardless of how weak the perturbations/interactions may be, whereas the n-based traditional test is good at detecting marginal associations. It is important that the two different approaches can work side by side, complementing each other.
The proposed method should have broad applications to other high-dimension (large p) -omics studies, such as epigenomic, transcriptomic, proteomic, metabolomic and exposomic studies, etc. It would be even better to have a cross-omics study, and/or with all its study subjects further linked to existing government or private-sector databases, such as, data of health insurances, traffic violations, internet usages, etc. A researcher conducting such a data-mining study has the potentials to push the p (the number of auxiliary/perturbation variables) to the millions, billions or even trillions and be rewarded with a very high power for detecting a weak association. Such a p-based method may set a stage for a new paradigm of statistical tests.
Methods
Crude null and sharp null in a case-control study
Let R = 1 indicate a subject is recruited in a study, R = 0, otherwise. In a case-control study, the recruitment process depends only on the disease status of a subject, that is,

Under the crude null of

we have

and therefore,

Under the sharp null of

we have

and therefore,

Testing crude null: n-based test
In a case-control study conducted in the study population, testing the crude null amounts to testing the equality of prevalence odds of X, between the case group ( ) and the control group (
) and the control group ( ), or equivalently, testing whether the odds ratio of X and D equals one:
), or equivalently, testing whether the odds ratio of X and D equals one:

Supplementary Table S1 presents the cell counts of a case-control study (ignore the variable, Z, for now). One may use the following test statistic:

 is distributed asymptotically as a chi-squared distribution with one degree of freedom (df) under the crude null.
is distributed asymptotically as a chi-squared distribution with one degree of freedom (df) under the crude null.
Power comparison
The power of the traditional n-based  is:
is:

where  is a df = 1 noncentral chi-squared distribution with noncentrality parameter,
is a df = 1 noncentral chi-squared distribution with noncentrality parameter,

Note that the power of  is determined by the significance level: α, the sample size: n (or more exactly the expected cell counts) and the effect size:
is determined by the significance level: α, the sample size: n (or more exactly the expected cell counts) and the effect size:

Assuming that a panel of independent auxiliary variables contains a certain proportion, π (0 ≤ π ≤ 1), of perturbative Zs such that  follows a normal distribution with a mean of zero and a variance of σ2 > 0 the theoretical power of the p-based MPT based on such panel is:
follows a normal distribution with a mean of zero and a variance of σ2 > 0 the theoretical power of the p-based MPT based on such panel is:

where

Note that in addition to α and n, the power of MPT is also determined by the total number of auxiliary variables: p and the ‘informativeness’ of the auxiliary variables:

(the product of perturbation proportion and perturbation strength).
We consider an X that is very weakly associated with D:

We also consider a panel of independent Zs. The logarithm of  follows a normal distribution with a mean of zero and a variance of 0.5 (a probability of 95% that an
follows a normal distribution with a mean of zero and a variance of 0.5 (a probability of 95% that an  is between 0.25 ~ 4.00). We consider four different values for the perturbation proportion (π = 1.0, 0.2, 0.1 and 0.05, respectively), with each perturbative Z having a weak perturbation effect (σ2 = 0.001, i.e., a probability of 95% that the ratio,
is between 0.25 ~ 4.00). We consider four different values for the perturbation proportion (π = 1.0, 0.2, 0.1 and 0.05, respectively), with each perturbative Z having a weak perturbation effect (σ2 = 0.001, i.e., a probability of 95% that the ratio,  , is between 0.94 ~ 1.06). The informativeness of Zs is therefore 0.001, 0.0002, 0.0001 and 0.00005, respectively. For convenience, the prevalence of X and each and every one of Zs is set at 40% for the control group. The significance level is set at α = 0.05.
, is between 0.94 ~ 1.06). The informativeness of Zs is therefore 0.001, 0.0002, 0.0001 and 0.00005, respectively. For convenience, the prevalence of X and each and every one of Zs is set at 40% for the control group. The significance level is set at α = 0.05.
Calculation of p-value using permutation
If the Zs in the panel are independent of one another, MPT is asymptotically a df = p chi-squared distribution under the sharp null. The critical value of MPT therefore is simply  when the level of significance is set at α. In actual practice however, Zs may not be independent of one another and sample size may be too small for an adequate chi-square approximation. Therefore, we need to rely on computer-intensive methods to simulate the null sampling distribution of MPT. With p = 1, Buzkova et al. pointed out that the method of parametric bootstrap is valid but the method of permutation (shuffling disease status between subjects) is conservative (overestimating the critical value)26. However, we found that as p increases, the permutation method remains slightly conservative but the parametric method becomes too liberal (underestimating the critical value). To err on the safe side, we therefore propose to use the permutation method to approximate the null sampling distribution of MPT.
when the level of significance is set at α. In actual practice however, Zs may not be independent of one another and sample size may be too small for an adequate chi-square approximation. Therefore, we need to rely on computer-intensive methods to simulate the null sampling distribution of MPT. With p = 1, Buzkova et al. pointed out that the method of parametric bootstrap is valid but the method of permutation (shuffling disease status between subjects) is conservative (overestimating the critical value)26. However, we found that as p increases, the permutation method remains slightly conservative but the parametric method becomes too liberal (underestimating the critical value). To err on the safe side, we therefore propose to use the permutation method to approximate the null sampling distribution of MPT.
Monte-Carlo simulation
We perform Monte-Carlo simulation to study the statistical properties of MPT empirically. The parameter setting is the same as the previous section. The sample size is set at n = 1,000. But to avoid the heavy computation burdens of simulating a very large panel of Zs, this time we let Zs have a perturbation proportion of 1.0 and a larger perturbation strength (σ2 = 0.004, a probability of 95% that  is between 0.88 ~ 1.13). Additionally, we also consider dependent Zs. Specifically, we simulate Zs using a first-order Markov chain, in both the case and the control groups, assuming an odds ratio between successive Zs of 2.0 (mild dependency) and 5.0 (strong dependency), respectively. We perform a total of 1,000 simulations. In each round of the simulation, we conduct 1,000 permutations to obtain an empirical P-value for MPT. The power of MPT is then calculated as the proportion of the simulations with a P-value < 0.05.
is between 0.88 ~ 1.13). Additionally, we also consider dependent Zs. Specifically, we simulate Zs using a first-order Markov chain, in both the case and the control groups, assuming an odds ratio between successive Zs of 2.0 (mild dependency) and 5.0 (strong dependency), respectively. We perform a total of 1,000 simulations. In each round of the simulation, we conduct 1,000 permutations to obtain an empirical P-value for MPT. The power of MPT is then calculated as the proportion of the simulations with a P-value < 0.05.
The type I error rates of MPT for panels of independent and dependent Zs (odds ratio between successive Zs = 5.0) are also empirically checked using Monte-Carlo simulations, for different number of subjects (n = 500, 1,000, 5,000) and number of auxiliary variables (p = 100, 1,000, 5,000). (Both n and p are assumed to be fixed by design.) Here X is a sharp null, that is, X has no effect on disease in any level stratified by Zs (no perturbation effect for all Zs: I = π × σ2 = 0). Other parameters are the same as in power simulations. We perform a total of 1,000 simulations, each round with 1,000 permutations.
Application to real data
MPT is applied to a public-domain data from a genome-wide association study of age-related macular degeneration6. The study recruited 146 individuals (96 cases and 50 controls) and genotyped 116,212 single nucleotide polymorphisms (SNPs). A total of 6,639 SNPs located on chromosome 1 (where previous studies27,28 have identified a number of significant susceptibility genes) with call rate > 95%, minor allele frequency > 5% and in Hardy-Weinberg equilibrium in the control group is included in the analysis. At each SNP, heterozygote and variant homozygote are grouped together.
In the analysis, each SNP takes turn to be the X and the remaining SNPs, the Zs. (The number of auxiliary variables is p = 6638, for each and every one of the total 6639 SNPs. This number is set prior to the MPT analysis to avoid complicating the multiple testing problem.) For a low-frequency SNP, some of the cells in Supplementary Table S1 may be empty. In that case, it is totally uninformative as a perturbation variable, because its  statistic is zero with the convention: 0 × log0 = 0. The P-value of the MPT for each SNP is obtained from 500,000 rounds of permutation. Because we repeatedly test each and every one of the 6639 SNPs for significance, for multiple testing correction the false discovery rate (FDR) is controlled at 0.05 using the q-values (QVALUE software)13. (Because of the dependence between SNPs, the q-value approach actually controls the FDR to be less than the nominal 0.0513,29,30.) Note that our fixation/drifting analysis does not create a multiple testing problem by itself, because the procedure was done only after the significance of a SNP had been determined.
statistic is zero with the convention: 0 × log0 = 0. The P-value of the MPT for each SNP is obtained from 500,000 rounds of permutation. Because we repeatedly test each and every one of the 6639 SNPs for significance, for multiple testing correction the false discovery rate (FDR) is controlled at 0.05 using the q-values (QVALUE software)13. (Because of the dependence between SNPs, the q-value approach actually controls the FDR to be less than the nominal 0.0513,29,30.) Note that our fixation/drifting analysis does not create a multiple testing problem by itself, because the procedure was done only after the significance of a SNP had been determined.
References
Siontis, G. C. & Ioannidis, J. P. Risk factors and interventions with statistically significant tiny effects. Int. J. Epidemiol. 40, 1292–1307 (2011).
Grontved, A. & Hu, F. B. Television viewing and risk of type 2 diabetes, cardiovascular disease and all-cause mortality: a meta-analysis. JAMA 305, 2448–2455 (2011).
Hemila, H. & Chalker, E. Vitamin C for preventing and treating the common cold. Cochrane Database Syst. Rev. 1, CD000980 (2013).
Ioannidis, J. P., Trikalinos, T. A. & Khoury, M. J. Implications of small effect sizes of individual genetic variants on the design and interpretation of genetic association studies of complex diseases. Am. J. Epidemiol. 164, 609–614 (2006).
Hindorff, L. A. et al. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U S A 106, 9362–9367 (2009).
Klein, R. J. et al. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–389 (2005).
Holliday, E. G. et al. Insights into the genetic architecture of early stage age-related macular degeneration: a genome-wide association study meta-analysis. PLoS One 8, e53830 (2013).
Fritsche, L. G. et al. Seven new loci associated with age-related macular degeneration. Nat. Genet. 45, 433–439, 439e1–439e2 (2013).
Wellcome Trust Case Control, C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Ollier, W., Sprosen, T. & Peakman, T. UK Biobank: from concept to reality. Pharmacogenomics 6, 639–646 (2005).
Chen, Z. et al. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int. J. Epidemiol. 40, 1652–1666 (2011).
Chapman, K., Ferreira, T., Morris, A., Asimit, J. & Zeggini, E. Defining the power limits of genome-wide association scan meta-analyses. Genet. Epidemiol. 35, 781–789 (2011).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U S A 100, 9440–9445 (2003).
Chatterjee, N., Kalaylioglu, Z., Moslehi, R., Peters, U. & Wacholder, S. Powerful multilocus tests of genetic association in the presence of gene-gene and gene-environment interactions. Am. J. Hum. Genet. 79, 1002–1016 (2006).
Gauderman, W. J., Murcray, C., Gilliland, F. & Conti, D. V. Testing association between disease and multiple SNPs in a candidate gene. Genet. Epidemiol. 31, 383–395 (2007).
Wang, T., Ho, G., Ye, K., Strickler, H. & Elston, R. C. A partial least-square approach for modeling gene-gene and gene-environment interactions when multiple markers are genotyped. Genet. Epidemiol. 33, 6–15 (2009).
Pan, W. Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genet. Epidemiol. 33, 497–507 (2009).
Pan, W. Statistical tests of genetic association in the presence of gene-gene and gene-environment interactions. Hum. Hered. 69, 131–142 (2010).
Bhutto, I. & Lutty, G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch's membrane/choriocapillaris complex. Mol. Aspects Med. 33, 295–317 (2012).
Ambati, J. & Fowler, B. J. Mechanisms of age-related macular degeneration. Neuron 75, 26–39 (2012).
Tsaur, M. L., Chou, C. C., Shih, Y. H. & Wang, H. L. Cloning, expression and CNS distribution of Kv4.3, an A-type K+ channel alpha subunit. FEBS Lett. 400, 215–220 (1997).
Lee, Y. C. et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann. Neurol. 72, 859–869 (2012).
Duarri, A. et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann. Neurol. 72, 870–880 (2012).
Banks, D. et al. L2DTL/CDT2 and PCNA interact with p53 and regulate p53 polyubiquitination and protein stability through MDM2 and CUL4A/DDB1 complexes. Cell Cycle 5, 1719–1729 (2006).
Bhattacharya, S., Chaum, E., Johnson, D. A. & Johnson, L. R. Age-related susceptibility to apoptosis in human retinal pigment epithelial cells is triggered by disruption of p53-Mdm2 association. Invest. Ophthalmol. Vis. Sci. 53, 8350–8366 (2012).
Buzkova, P., Lumley, T. & Rice, K. Permutation and parametric bootstrap tests for gene-gene and gene-environment interactions. Ann. Hum. Genet. 75, 36–45 (2011).
Lim, L. S., Mitchell, P., Seddon, J. M., Holz, F. G. & Wong, T. Y. Age-related macular degeneration. Lancet 379, 1728–1738 (2012).
Gorin, M. B. Genetic insights into age-related macular degeneration: controversies addressing risk, causality and therapeutics. Mol. Aspects Med. 33, 467–486 (2012).
Storey, J. D. The positive false discovery rate: a Bayesian interpretation and the q-value. Ann. Stat. 31, 2013–2035 (2003).
Storey, J. D., Taylor, J. E. & Siegmund, D. Strong control, conservative point estimation and simultaneous conservative consistency of false discovery rates: a unified approach. J. R. Stat. Soc. B. 66, 187–205 (2004).
Acknowledgements
This paper is partly supported by grants from Ministry of Science and Technology, Taiwan (NSC 102-2628-B-002-036-MY3) and National Taiwan University, Taiwan (NTU-CESRP-102R7622-8). No additional external funding received for this study. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
W.-C.L. designed the study. M.-T.L. performed simulations, analyzed the data and prepared tables and figures. W.-C.L. and M.-T.L. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images in this article are included in the article's Creative Commons license, unless indicated otherwise in the image credit; if the image is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the image. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Lo, MT., Lee, WC. Detecting a Weak Association by Testing its Multiple Perturbations: a Data Mining Approach. Sci Rep 4, 5081 (2014). https://doi.org/10.1038/srep05081
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05081
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
