
Abstract
Prolificacy can directly impact porcine profitability, but large genetic variation and low heritability have been found regarding litter size among porcine breeds. To identify key differences in gene expression associated to swine reproductive efficiency, we performed a transcriptome analysis of sows’ endometrium from an Iberian x Meishan F2 population at day 30–32 of gestation, classified according to their estimated breeding value (EBV) as high (H, EBV > 0) and low (L, EBV < 0) prolificacy phenotypes. For each sample, mRNA and small RNA libraries were RNA-sequenced, identifying 141 genes and 10 miRNAs differentially expressed between H and L groups. We selected four miRNAs based on their role in reproduction and five genes displaying the highest differences and a positive mapping into known reproductive QTLs for RT-qPCR validation on the whole extreme population. Significant differences were validated for genes: PTGS2 (p = 0.03; H/L ratio = 3.50), PTHLH (p = 0.03; H/L ratio = 3.69), MMP8 (p = 0.01; H/L ratio = 4.41) and SCNN1G (p = 0.04; H/L ratio = 3.42). Although selected miRNAs showed similar expression levels between H and L groups, significant correlation was found between the expression level of ssc-miR-133a (p < 0.01) and ssc-miR-92a (p < 0.01) and validated genes. These results provide a better understanding of the genetic architecture of prolificacy-related traits and embryo implantation failure in pigs.
Similar content being viewed by others


Introduction
Pig is economically one of the most important species. Reproductive traits such as fertility and prolificacy can directly impact porcine profitability, becoming one of the most relevant traits from a genetic and economic point of view. The annual production of a sow is determined to a large degree by its litter size in terms of total number of piglets born (TNB) and number of piglets born alive (NBA) per parity. Total number of piglets born and NBA are the most important reproductive traits used in swine breeding programmes1.
Although sow’s fertility depends directly on the ovulation rate (OR), litter size is not strongly determined by this factor, but by the capacity of maintaining viable embryos throughout gestation. Prenatal mortality could be a determinant factor for litter size in pigs2,3. The relevance and timing of embryonic and foetal losses during gestation have been reported in many studies and it is estimated that about 25–45% of fertilized ova do not survive through gestation. Losses of embryos and foetuses occur at each stage of development and are primarily determined by the uterine capacity of the pregnant sows4. A large genetic variation has been found among porcine breeds regarding litter size, being the Chinese Meishan one of the most prolific pig breeds known5.
Improvements in litter size across the swine industry have occurred through different selection schemes such as phenotypic, family index, best linear unbiased prediction or hyper-prolific-based selection methods2. Being a complex trait regulated by a large number of genes, along with its low heritability, has made the selection of this character rather challenging for a number of years6. To date, main used strategies to detect those genes affecting litter size and its components have been: linkage analyses based on the identification of genomic regions linked with a phenotypic reproduction trait and candidate gene approaches, based on a priori knowledge of a gene having a high probability to play a relevant role in reproduction by their physiological role or location7.
Significant quantitative trait loci (QTL) associated with porcine reproductive traits have been identified in our study population and many others: SSC3, SSC8, SSC9, SSC10 and SSC15 for ovulation rates8,9,10,11, SSC7, SSC8, SSC12, SSC13, SSC14 and SSC17 for total number piglets born6,12,13, SSC4 and SSC13 for number of stillborn10,14 and SSC8 for uterine capacity and prenatal survival13,15. Although there are even more QTLs reported for litter size component traits, most of these results are inconsistent and true causal genes still remain scant due to the large disequilibrium linkage blocks present in the genome of livestock species16.
In recent years, the knowledge obtained by deciphering the pig genome and advances in molecular genetics, such as the transcriptomic analysis by RNA sequencing, have provided a powerful tool to better understand the genetic architecture of prolificacy-related traits. Recent years have seen a remarkable rise in porcine transcriptomic data. The use of microarrays and large-scale transcriptome analysis to identify differentially expressed genes in specific tissues, cell types or breeds has shed light on many aspects of porcine production traits17,18,19,20,21,22,23,24. Despite this, there have only been a few comparative studies on uterine function for prolific pigs and a low number of experiments regarding differences in endometrial gene expression between porcine breeds25,26,27.
In swine, during the oestrus cicle and throughout pregnancy many critical morphological and secretory changes take place in the uterus. These sets of physiological changes are clear evidence of the extremely complex interactions taking place between gene products and of remarkable transcriptomic reorganization. This highlights the importance of performing profiling experiments in porcine breeds with extreme prolificacy phenotypes, in order to better understand those gene interactions and the regulatory mechanisms affecting litter size in pigs.
An important mechanism of gene expression regulation is miRNAs. It is well known that miRNAs have key functions in many relevant biological processes, including cellular differentiation, proliferation and apoptosis28. All these processes are involved in embryo formation, early development and implantation. Although the exact role of miRNAs in normal embryo formation and endometrial preparation for pregnancy still remains unclear, they have been widely associated with mammalian development29. Moreover, Yu et al., demonstrated that miRNA expression in mouse embryos was higher than in mature mouse tissues, confirming their role during embryo development30.
The goal of our study is, then, to define those genes and miRNAs that are differentially expressed in the uterine endometrium of pregnant sows with extreme prolificacy phenotypes in an Iberian x Meishan F2 population. These two porcine breeds differ significantly in their prolificacy levels, being the Meishan breed one of the most prolific porcine breeds, with an average of 14.3 piglets born alive per parity31, whereas the Iberian breed is considered a very low-prolificacy breed with an average of 7 piglets per parity32. This makes our study population highly suitable for further investigating the biological underpinnings that contribute to controlling litter size in pigs.
Results
Differential gene expression
Uterine receptivity to implantation is a process that can be very different, depending on the species, but always involves several changes in the expression of genes that are directly involved in pathways, such as progesterone and oestrogen biosynthesis, immune recognition, membrane permeability, angiogenesis and vasculogenesis, transport of nutrients and signalling for pregnancy recognition. Thus, changes in the expression level of those genes may influence uterine receptivity to implantation. Analysis of read counts revealed a total of 141 differentially expressed genes (DEG) between high- and low-prolificacy samples when a false discovery rate (FDR) corrected q-value of 0.05 was set as the threshold for significance (supplementary table S1). Expression differences between H and L groups ranged from 5.61 to −5.84 fold. A total of 55 transcripts showed an overexpression in the high-prolificacy group, with expression differences ranging from −1.45 to −5.84 fold, whereas 49 showed an overexpression in the low-prolificacy group, with expression changes ranging from 1.51 to 5.61 fold. Moreover, we identified 27 transcripts expressed uniquely in the L group (2 annotated genes and 25 unannotated transcripts) and 10 transcripts expressed uniquely in the H group, including 4 annotated genes and 6 unannotated transcripts (See supplementary table S2).
Functional annotation and QTL mapping analysis
In order to establish whether differentially expressed genes found were involved in a relevant biological process for any stage of pregnancy establishment and development in the pig, we performed a gene ontology (GO) annotation and enrichment analysis. Obtained results revealed that the top over-represented functions were related with female pregnancy (q-value = 0.0001), maternal placenta development (q-value = 0.024) and decidualization (q-value = 0.024). All p-values were estimated through Chi square analysis and FDR corrected. An FDR-corrected q-value of 0.10 was set as the threshold for significant functional enrichment (See table 1). We also performed this enrichment analysis considering separately those genes overexpressed in either group. The DEG overexpressed in H prolificacy samples were clustered in seven enriched general biological processes, including mainly: positive regulation of cell proliferation (GO: 0008284; q-value = 3.67E-06) and response to hypoxia (GO: 0001666; q-value = 0.0002). Differentially expressed genes showing an overexpression in L prolificacy samples were clustered in 11 enriched general biological processes, including mainly: proteolysis/cell-cell signalling (GO: 0006508, GO: 0007267; q-value = 2.36E-06) and in utero embryonic development (GO: 0001701; q-value = 0.0001).
In order to focus on those genes that could be strongly associated with reproduction and have an impact on litter size variation, a chromosomal localization of DEGs within known QTL intervals was performed. We identified a total of 59 mapping into known reproductive QTLs. Among them, 25 were located within a QTL specifically related with litter size: total number of piglets born alive (NBA), total number of piglets born (TNB), total number of piglets stillborn (TSB), body weight at birth (BW), body weight at 10 weeks (WT), body weight at weaning (WWT), mummified pigs (MMUM) and/or ovulation rate (OVRATE). Results are shown in supplementary table S3.
Candidate genes selection and expression levels validation: RT-qPCR
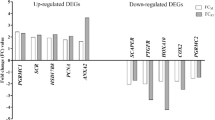
Among the 141 genes found differentially expressed in the RNAseq analysis (q-value < 0.05), we selected those displaying the most extreme differences between H and L groups (fold change ≥ 3) reducing the initial set to 28 genes. Based on the results obtained after the QTL mapping, we considered only those that have a positive mapping into known reproductive QTLs, reducing this number to 14 genes. Finally, considering the gene ontology (GO) annotation and enrichment analysis results and based on their known role in any relevant pathway related with reproduction, pregnancy or embryonic development, we chose 5 candidates: HPGD, MMP8, PTGS2, PTHLH and SCNN1G (See Table 2). Expression data obtained by RNA sequencing for these candidate genes was validated by RT-qPCR in 36 extreme individuals (H, n = 18; L, n = 18) of our F2 population. We confirmed significant differences in the expression level of four of these five genes between H and L samples with an H/L ratio > 3.5: MMP8 (mean H = 0.174, mean L = 0.035; p-value = 0.011), PTGS2 (mean H = 0.144, mean L = 0.038; p-value = 0.026), PTHLH (mean H = 0.126, mean L = 0.033; p-value = 0.034) and SCNN1G (mean H = 0.117, mean L = 0.031; p-value = 0.048). Results are shown in Fig. 1a. The observed ratios between the expression level of selected candidate genes were similar in our RNAseq and RT-qPCR analysis: HPGD (RNAseq FC = 1.85, RT-qPCR FC = 1.81), PTGS2 (RNAseq FC = 4.06, RT-qPCR FC = 3.79), PTHLH (RNAseq FC = 4.32, RT-qPCR FC = 3.78) and SCNN1G (RNAseq FC = 3.65, RT-qPCR FC = 3.72). Only for the MMP8 gene the observed ratios between both analysis were slightly different (RNAseq FC = 2.99, RT-qPCR FC = 4.92).

(a) RT-qPCR analysis results for gene expression. Expression values were calculated applying the −2∆∆CT algorithm. Estimated relative quantities were normalized for the expression value of two uterus endogenous genes B2MG and UBC and calibrated to the sample with a higher expression. Significance was set at a p-value < 0.05 (*). (b) RT-qPCR analysis results for miRNA expression. Relative quantities were calculated using target-specific amplification efficiencies and normalized for the expression level of two uterus reference miRNAs: has-miR-93 (M = 0.464; CV = 0.156 and ssc-miR-103 (M = 0.464; CV = 0.166).
Differential miRNA expression and in silico target prediction
The observed differences in the expression level of these genes between H and L prolificacy groups suggests that a different regulation mechanism may be occurring. We hypothesize that known gene regulators such as miRNAs could be responsible for this. Sequencing analysis revealed a total of 341 miRNAs being expressed in H and 329 in L prolificacy samples. Among all expressed microRNAs found in our endometrial samples, a total of 10 mature miRNAs were predicted as differentially expressed between H and L prolificacy phenotypes when considering a p-value < 0.05. However, we lost this significance when applying the same FDR correction significance criteria as used for DEG identification (Supplementary table S4).
To explore the possible regulatory role of these differentially expressed miRNAs, we predicted their potential target genes using TargetScan software. Five of these 10 differentially expressed miRNAs had as a putative mRNA target one of the DEGs found between the H and L groups (Supplementary table S5). The novel prediction tool from the mirDeep package allowed us to also identify 15 putative novel miRNAs in H samples and 12 in L samples, with an estimated probability of being a genuine miRNA precursor greater than 90% (Supplementary table S6).
Candidate miRNAs selection and expression levels validation: RT-qPCR
Among the 10 miRNAs found differentially expressed in the RNAseq analysis (q-value < 0.05), we selected as candidates those that have been extensively reported in the literature as relevant in the regulation of reproduction-related genes in both pig and human: ssc-miR-92a, ssc-miR-101, ssc-miR-133a and ssc-miR-181d (See Table 3). We validated their expression levels by RT-qPCR in the same 36 F2 extreme individuals (H, n = 18; L, n = 18) used for gene expression validations (Table 3).
Obtained results revealed similar expression levels between both prolificacy groups for these four miRNAs (Fig. 1b). However, significant correlations were found between the expression level of prolificacy-related miRNAs ssc-miR-92a and ssc-miR-133a and validated DEG analysed by RT-qPCR (Table 4). Again, the observed fold changes were similar in both analysis: ssc-miR-92a (RNAseq FC = 1.26, RT-qPCR FC = 1.09), ssc-miR-101 (RNAseq FC = 1.20, RT-qPCR FC = 0.94), ssc-miR-181d-5p (RNAseq FC = 1.16, RT-qPCR FC = 0.95). This confers consistency to our findings and led us to think that the observed differences in the expression levels between H and L groups represent the real biological background of our samples.
Biological role of candidate genes: Interactions and upstream regulators
To place the results in a biological context that allows us to better understand them, we performed an Ingenuity Pathway Analysis (IPA) to analyze the existing networks and potential molecular interactions between the validated candidate genes. Along pregnancy, hormones and other molecules secreted from the porcine conceptus act directly on the endometrium promoting its interaction with maternal uterus and placental development. We identified multiple links and interactions between our validated candidate genes and some molecular components. In the predicted network generated by IPA algorithm (Fig. 2), we observed that the expression of our four validated candidates could be modulated mainly by three molecules: trypsin (for genes MMP8, PTGS2 and SCNN1G), insulin (for gene SCNN1G) and the vascular endothelial growth factor (Vegf) which acts on PTHLH gene.

Ingenuity Pathway Analysis (IPA) Core Analysis-based network.
Links of validated genes and other genes or molecules are represented with a continuous (direct interaction) or discontinuous line (indirect interaction).
After performing the analysis of the putative common upstream regulators we identified that the common regulators to all four genes are the cytokines Interleukin 1 beta (ILK-1β, p-value = 0.000007) and the tumor necrosis factor ligand (TNF, p-value = 0.00008). Results are shown in Table 5.
Discussion
In this study, we investigated the whole transcriptome profile of the swine endometrial epithelium in an Iberian x Meishan F2 population using RNA sequencing (RNA-seq), with the aim to identify key differences in gene expression associated to swine reproductive efficiency. Understanding the complexity of the key mechanisms for successful reproduction in humans and animals has been challenging. Even though a few studies have addressed this goal, this study represents one of the first descriptions of the mechanisms that affect embryonic survival in the pig, providing the knowledge to enhance fertility and reproductive health in this species.
The main limitation of increasing litter size in pigs is prenatal mortality. Two critical stages are early and mid-gestation, responsible for around 20–30% (days 10–30 of gestation) and 10–15% (days 50–70 of gestation) of embryonic loss respectively2. Recent evidences have indicated that the prenatal loss in pigs results mainly from the decreased placental efficiency and uterine capacity33,34.
Uterine receptivity to implantation is a process that can be very different, depending on the species, but always involves several changes in the expression of genes that are directly involved in pathways, such as progesterone and oestrogen biosynthesis, immune recognition, membrane permeability, angiogenesis and vasculogenesis, transport of nutrients and signalling for pregnancy recognition35,36. Thus, changes in the expression level of those genes may influence uterine receptivity to implantation.
In this study we have identified 141 differentially expressed genes between high and low prolificacy samples. Functional enrichment analysis suggested that most of these genes are directly involved in the above-mentioned biological processes, which are highly relevant for pregnancy and some specific stages of embryonic development in swine. We have focused our validations on a first set of genes that are up-regulated in our high-prolificacy samples. Some of those genes are also located inside the confidence intervals of previously described reproduction QTLs: ovulation rate, gestation length, number of piglets born alive and embryo’s birth weight. Considering these, we proceeded to validate their expression by real time RT-qPCR. As predicted in the RNAseq analysis, four of these genes were differentially expressed in our endometrial samples, being overexpressed in those with a high-prolificacy phenotype.
Several DEGs found in our samples have been extensively discussed by many authors before37,38,39,40,41,42 and their involvement in the establishment of pregnancy and in the physiological, molecular and structural changes that take place in the uterine tissue to promote embryo implantation have been demonstrated in pigs and other mammals . Their involvement in many stages of embryonic development postulate them as key factors for deciphering the mechanisms involved in the regulation of litter size in our study population.
Prostaglandins (PGs) produced by the uterus play an important role in regulation of the oestrous cycle and during early pregnancy in pigs and many other species43. In the porcine endometrium, luteoprotective PGE2 and luteolytic PGF2α are the main PGs produced and pregnancy establishment depends directly in a proper ratio between the synthesis of both. An inhibition of PG synthesis results in pregnancy failure44. One of the validated genes found differentially expressed among our samples is the prostaglandin endoperoxide synthase (PTGS; also known as prostaglandin G/H synthase or cyclooxygenase COX2). The PTGS2 gene has been widely discussed over the years and its key function to ensure reproductive success has been widely demonstrated through several previous studies. It constitutes a rate-limiting enzyme in the production of PGs as it catalyzes the conversion of arachidonic acid to PGH2, which is a common substrate for various prostaglandins. Its conserved role in implantation in various species, including humans, has previously been discussed45,46. Thus, considering that the production of prostaglandins directly contributes to the successful establishment of pregnancy and that uterine receptivity to implantation is progesterone-dependent, a lack in the expression of this gene will directly affect the appropriate conceptus attachment. It has been observed that the expression of PTGS1 and PTGS2 is substantially increased during implantation. We speculate that the underexpression of this gene in our low-prolificacy samples may contribute to embryonic deaths due to deficiencies in progesterone synthesis. This uterine receptivity via expression of PTGS2 gene is a process that has been demonstrated to be directly regulated by another key gene also found DE in our samples: KLF5. This gene belongs to the Kruppel-like factors (KLFs) family. This is a zinc finger-containing transcription factor, which is known to regulate several cellular processes, including development, differentiation, proliferation and apoptosis47. At the beginning of the attachment reaction, the first cell type to interact with the blastocyst trophectoderm is the uterine luminal epithelium. KLF5 function is critical to make this uterine luminal epithelium conducive to blastocyst implantation and growth. In its absence, trophectoderm development is defective, resulting in developmental arrest at the blastocyst stage48. These results suggest that KLF5 is a key regulator of embryo pre-implantation49. Thus, the fact that this gene is overexpressed in our high-prolificacy samples strengthens our idea of the important effect it may have on prolificacy levels and litter size control.
As mentioned before, successful establishment of pregnancy also depends on many structural changes that take place in the uterine tissue. Species with invasive implantation require a cell-to-cell communication through connexin proteins. Although porcine implantation is superficial, some authors have reported that endometrial cell-to-cell interaction may also be necessary for limiting trophoblast invasiveness or to develop specific channels that allow this superficial implantation50. And it is at this stage where the validated gene MMP8 plays a key role. Proteins such as matrix metalloproteinase (MMP) are a family of enzymes (with more than 20 members identified) that use zinc-dependent catalysis to break down the components of the extracellular matrix (ECM)39,51,52. We hypothesize that the observed significant overexpression of this gene in our high-prolificacy samples may indicate a more efficient tissue reorganization to support the growing foetus.
Another relevant structural gene found differentially expressed in our extreme F2 population is the Forkhead transcription factor FOXA2. FOXA transcription factors are a subfamily of Forkhead transcription factors that has been found to play an important role in early development, organogenesis, metabolism and homeostasis53. Low-prolificacy samples show a decreased expression of this gene compared to those with high prolificacy, supporting our idea that an underexpression of this gene could be leading to defects in early development, affecting stages such as gastrulation or, later on, in embryo morphogenesis.
Many other genes found differentially expressed in this study are closely related with critical stages in embryo development at implantation level or later in the survival of the embryo itself. This has provided us with a powerful list of candidates that require further validations in order to prove their direct involvement in the control of litter size in swine. Because of the usefulness of the pig as a biomedical model and the parallelism in the function of these genes in humans, this study also provides a powerful tool to understand which genes are key in the process of embryo survival in mammals.
We also wanted to explore the regulatory mechanisms that do mediate this differential expression in our study population. To do so, we have also analysed the miRNA expression profile in both extreme phenotypic groups.
We predicted a differential expression of 10 mature miRNAs between our H and L prolificacy samples. Some of these differentially expressed miRNAs have been demonstrated to be directly involved in the regulation of reproductive-related genes in pig and other mammals54,55,56,57. After this preliminary bioinformatic screening we proceeded to the experimental validation of the expression level of 4 of these 10 miRNAs, considering their role in reproductive-related pathways: ssc-miR-92a, ssc-miR-101, ssc-miR-133a and ssc-miR-181d.
In concordance with RNAseq predictions, ssc-miR-101, ssc-miR-133a and ssc-miR-181d were overexpressed in L samples while ssc-miR-92a was overexpressed in H samples. MiR-92, belongs to the miR-17 ~ 92 cluster, demonstrated in recent reports to regulate cardiac development, endothelial cell proliferation and angiogenesis, which are relevant processes for embryogenesis and pregnancy itself58. Loss and gain of function experiments showed that miR-92a inhibited angiogenesis in vitro and in vivo59 and that deletion of miR-92a is sufficient to induce a developmental skeletal defect55. Thus, the observed overexpression of this miRNA in our H samples could be explained by its positive effect on several key processes for pregnancy and embryo development.
Real-time RT-qPCR analysis revealed similar expression levels of these miRNAs in both groups (FC < 1.5). However, it has been demonstrated that even very small changes in microRNA expression levels (FC 1.5 to 2.5) could have a direct impact on their target genes and some authors have observed these small differences when performing miRNA differential expression studies related to reproductive processes60,61. We hypothesize that this could be caused by an insufficient sequencing depth in our libraries, because despite these similar miRNA expression levels observed between both phenotypes, a significant correlation was found between the expression levels of validated genes PTHLH, MMP8, PTGS2 and SCNN1G and both ssc-miR-133a and ssc-miR-92a. Therefore, the finding of this significant correlation leads us to think that the observed differences, despite being low, may be biologically significant. Many years ago, Calin et al. suggested that the capability of miRNAs to regulate multiple targets within the same pathway could amplify their biological effects62.
Besides miRNAs, upstream regulators such as transcription factors (TFs), growth factors (GFs) and many other molecules may play a critical role as drivers or master regulators of gene expression. Investigating their involvement in a particular gene network or pathway can provide better clues on the underlying regulatory mechanisms that do mediate the observed differences in the expression of key genes in a particular biological context.
In this study we have explored the regulatory role that some candidate miRNAs exert in the expression of key reproductive-related genes and the possible effect that this has on litter size control. In addition, we have established which interactions exist between our validated candidate genes and other known regulatory molecules. There are two cytokines particularly capable of acting on the expression of these four genes which are the ILK-1β and the TNF.
In reproductive biology, the role of these cytokines has been implicated in ovulation, menstruation and embryo implantation and pathological processes such as preterm delivery and endometriosis63,64. The interleukin 1 is a pro-inflammatory cytokine with multiple functions in a range of tissues65. All components of the IL-1 system have been examined in the human endometrium and have been implicated as an important mediator of embryo implantation66,67. Simón C. and collaborators, demonstrated in mice, that IL-1 receptor antagonist given before implantation significantly reduces the number of implanted embryos, indicating a role for IL-1 in embryo implantation64.
The TNF is a pro-inflammatory cytokine that plays an important role in modulating the acute phase reaction. It was first discovered in amnion and placenta68, but many studies have demonstrated the presence of this cytokine and its receptors in the diverse human reproductive tissues69. The TNF has been implicated in ovulation, corpus luteum formation and luteolysis and it has been related to many endometrial and gestational diseases such as amniotic infections, recurrent spontaneous abortions, preeclampsia, preterm labour or endometriosis70,71,72. Although these cytokines may be acting on the expression of our validated candidate genes, we haven’t seen them differentially expressed between H and L groups.
It is clear, that there is a complex network of interacting genes regulating litter size in pigs. However, this work has led to the identification of several potential candidate genes associated with critical steps involved in embryonic survival during the sow’s gestation. Our results also describe the possible regulatory mechanisms that could be responsible of the differences in the expression level of key genes related with litter size control in pigs.
Materials and Methods
Animal material and sample collection
Animals used in this study come from an F2 population resulting from the cross of 3 Iberian males from the Guadyerbas line (Dehesón del Encinar, Toledo, Spain) with 18 Meishan females (Domaine du Magneraud, INRA, France). Once the F1 generation was obtained, 8 boars and 97 sows were mated to obtain a 255 F2 progeny at the Nova Genètica S.A. experimental farm (Lleida, Spain).
During four consecutive parities, main parameters based on the sows’ reproductive efficiency were recorded: number of piglets born alive (NBA) and total number of piglets born (TNB) means. At day 30–32 of their fifth gestation, when litter size has reached the maximum73, sows were slaughtered and the number of corpora lutea (CL or OR) and number of foetuses (NF) attached to the uterus were also recorded. At slaughter, endometrial samples from the apical uterus of F2 sows were collected and subsequently snap-frozen in liquid nitrogen. Preservation and storage was made at −80 °C until usage. All animal procedures were carried out according to the European animal experimentation ethics law and approved by the institutional animal ethics committee of Institut de Recerca i Tecnologia Agroalimentàries (IRTA).
Phenotypic records and samples selection
F2 sows were ranked by their estimated breeding value (EBV), which was calculated by using best linear unbiased predictors (BLUP) according to the reproductive traits described above: NBA and TNB means, OR and NF. Based on this ranking, individuals were divided into two groups: high (H; EBV > 0) and low (L; EBV < 0) prolificacy. Among the whole F2 progeny (n = 255), individuals displaying the most extreme EBVs were selected to be used in this study (n = 36). All phenotypic records are shown in Table 6.
RNA isolation and quality assessment
Total RNA was extracted from sows’ endometrial samples using TRIzol® reagent (Invitrogen, Carlsbad, USA), following the manufacturer’s instructions. The RNA integrity was assessed using an Eukaryote Total RNA Nano 6000 Labchip on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, USA) and quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, USA). Only those RNA samples with an RNA integrity number (RIN) ≥ 7 were used in subsequent experiments.
Ion Torrent PGM libraries preparation and RNA sequencing
Ion Torrent adapter-ligated libraries were prepared from extracted total RNA according to the Ion Total RNA-seq Kit v2 protocol (Life Technologies – Part #4476286 Rev. B) following the manufacturer’s instructions.
mRNA libraries preparation
Samples corresponding to animals displaying very extreme EBVs and very high RNA quality (RIN≥ 8) were used to prepare mRNA libraries (H, n = 3; L, n = 3). We constructed sequencing libraries starting from 500 ng of total RNA. PolyA RNA fraction was purified from total RNA samples using the Dynabeads® mRNA DIRECT Micro Kit (Life Technologies – Part #1148804 Rev. A) following the manufacturer’s instructions. Each sample was subjected to Ion semiconductor sequencing using a 318 chip on an Ion-Torrent PGM sequencer.
Small RNA libraries preparation
Small RNA sequencing was also performed using 318 chips on an Ion-Torrent PGM sequencer. In this case, we used stored GS FLX 454 microRNA sequencing libraries that we had previously used in our research61, which included the same extreme samples used in the mRNA libraries protocol (H, n = 7; L, n = 5). To adapt these performed libraries to the Ion semiconductor sequencing technology protocol, it was necessary to remove the 454 specific adaptors and to add the Ion Torrent A and P1 specific ligators. After doing so, each miRNA library was re-sequenced.
Bioinformatics and statistical analysis
Approximately 5 million short single-end reads (≈200 bp) were obtained for each library and sample and were subsequently assembled into a non-redundant set of 30,585 gene transcripts (3,024,658,544 bp) from the available Sus scrofa genome alignment version 10.2 (available at http://www.ncbi.nlm.nih.gov/assembly/GCF_000003025.5/#/def). In average, 75% of the reads were successfully mapped to the Sus scrofa genome.
Quality control for single-end raw reads
Raw reads formatted as fastq files were processed using FastQC 0.10.1 (freely available at http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Considered low quality reads by applying FastQC defaults, were removed and all downstream analyses were performed only on those reads meeting the quality criteria. Ion Torrent A and P1 adaptors were removed using Cutadap 1.4 (freely available at http://code.google.com/p/cutadapt/).
Reads mapping, alignment and annotation
Obtained sequence reads from mRNA libraries were mapped with Tophat (v1.4.0) to the latest porcine genome sequence assembly (Sscrofa10.2, August 2011). Transcript isoforms were assembled using Cufflinks 2.1.1 and combined with gene annotations extracted from Ensembl (ftp://ftp.ensembl.org/pub/release-75/gtf/sus_scrofa). The criteria used to filter out unique sequence reads was: minimum length fraction of 0.9; minimum similarity fraction of 0.8 and a maximum number of 2 mismatches.
Sequence reads from small RNA libraries were analysed following the Perl scripts contained in the miRDeep 2.0 package74 (freely available at http://www.mdc-berlin.de/rajewsky/miRDeep). Briefly, reads were first collapsed to ensure that each sequence only occurs once. Collapsed reads were then mapped to predefined miRNA precursor sequences from the miRBase v.20 contained in the porcine genome sequence assembly (Sscrofa 10.2, August 2011). Finally, unmapped reads served as input sequences for the novel miRNAs prediction algorithm.
Differential gene expression, functional annotation and QTL mapping analysis
Analysis of differential gene expression across high and low-prolificacy groups was performed using Cuffdiff 2.0.2 which is included in the Cufflinks package (available at http://cufflinks.cbcb.umd.edu/manual.html). For small RNA libraries, differentially expressed miRNA genes were detected by using the DEseq R package 1.8.375. A Benjamini-Hochberg FDR corrected p-value of 0.05 was set as the threshold for significant differential expression in both cases.
Babelomics 4.3.0 (http://babelomics.bioinfo.cipf.es) was used to functionally annotate DEG. The pig functional annotation database is not as complete as human, therefore, we converted the pig gene IDs (Ensembl Sus scrofa 10.2) into human gene IDs using Ensembl BioMart tool (http://www.ensembl.org/biomart/martview/). Then the homologous human Ensembl IDs were submitted to the Babelomics database for functional annotation. P-values to estimate over-represented GO terms were obtained through Chi square analysis. An FDR-corrected p-value of 0.10 was set as the threshold for significance.
All differentially expressed genes found were mapped against the latest release (Aug 25, 2014) of the Pig Quantitative Trait Locus Database76. Those DEGs displaying a significant functional annotation related to reproduction processes and/or a positive mapping into known reproductive QTLs were selected as a first set of candidates for quantitative real-time PCR validations.
Expression level validation by reverse transcription quantitative real-time PCR (RT-qPCR)
Five candidate genes and four candidate miRNAs displaying significant differences in their expression level between H and L samples were validated by RT-qPCR. The same samples selected for RNA-seq were used in these validations, but in order to obtain a broader view of the expression level of these genes in our population, the sample size was expanded using other extreme F2 samples (H, n = 18; L, n = 18).
Reverse transcription (RT): cDNA synthesis
Extracted total RNA was quantified using an ND 1000 Nanodrop® Spectrophotometer (Thermo Scientific, Wilmington, USA). The RNA quality and integrity were determined using an Eukaryote Total RNA Nano 6000 Labchip on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, USA).
Synthesis of cDNA for gene expression validation was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) from 1 μg of total RNA in 20 μl reaction. The synthesis of cDNA for miRNA expression validation was performed using extracted total RNA as described by Balcells et al.77 Briefly, 600 ng of total RNA in a final volume of 20 μL including 10x poly (A) polymerase buffer, 0.1 mM of ATP, 0.1 mM of each dNTP, 1 μM of RT-primer, 200 U of M-MuLV Reverse Transcriptase (New England Biolabs, USA) and 2 U of poly (A) polymerase (New England Biolabs, USA) was incubated at 42 °C for 1 hour and 95 °C for 5 minutes for enzyme inactivation. The used RT-primer sequence was 5′-CAGGTCCAGTTTTTTTTTTTTTTTVN, where V is A, C and G and N is A, C, G and T. Minus reverse transcription (RT) and minus poly A) polymerase controls were performed.
Real-time RT-qPCR reaction
DE genes expression validation
Quantitative PCR reactions were performed in triplicate in 20 μL final volume including 10 μL SYBR® Select Master Mix (Life Technologies – Thermo Fisher Scientific, Massachusetts, USA), 300 nM of each primer and 5 μL of a 1:200 dilution of the cDNA. A 1:5 relative standard curve generated from a pool of equal amounts of cDNA from all samples was included in each qPCR assay to estimate qPCR efficiency. Reactions were incubated in a 96-well plate at 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 sec and 60 °C for 1 min on a 7900 HT Real-Time PCR System using 7900HT SDS v2.4 software (Applied Biosystems, USA). DNA primers for each gene were designed using Primer Express® software v2.0 (Applied Biosystems, USA) following manufacturer’s instructions (Table 7). Melting curve analysis was included in each qPCR to detect unspecific amplifications. Expression values were calculated with qbasePLUS software (Biogazelle) applying the −2∆∆Ct algorithm, after verifying that the assumptions of the method were met78. Estimated relative quantities were calibrated to the sample with a higher expression and normalized for the expression value of two uterus endogenous genes: B2MG79 and UBC80. Reference genes stability was also assessed with qBasePLUS software considering a GeNorm M value < 0.5 and a coefficient of variation (CV) < 0.2. Significance was set at a p-value < 0.05.
DE miRNAs expression validation and putative targets prediction
Quantitative PCR reactions were performed as described above but using a different concentration of primers according to each miRNA. DNA primers were designed following the methodology suggested by Balcells et al. (Table 8). Relative standard curves were included in each qPCR assay to estimate target-specific amplification efficiencies. Expression values were calculated with qbasePLUS software using these amplification efficiencies. Relative quantities were normalized for the expression value of two uterus reference miRNAs: has-miR-93 and ssc-miR-10381 and calibrated to the sample with a higher expression. Reference miRNAs stability was determined considering a GeNorm M value < 0.5 and a coefficient of variation (CV) < 0.2. Significance was set at a p-value < 0.05.
Biological putative targets prediction was performed using TargetScan 6.2 online software. Targets were considered true positives if conserved 8mer and 7mer sites match the seed region of each miRNA.
Analysis of candidate genes interactions and upstream regulators
The four validated genes (MMP8, PTHLH, PTGS2 and SCNN1G) were submitted to Ingenuity Pathway Analysis (IPA 4.0, Ingenuity Systems Inc., www.ingenuity.com) for mapping to canonical pathways and identifying upstream regulators. As the Ingenuity Knowledge Base relies on ortholog information for only human, mouse and rat, we submitted to IPA the correspondent human Ensembl IDs of our candidate genes. We ran the Core Analysis function designating a set of criteria: genes and endogenous chemicals, direct and indirect interactions, maximum molecules per network (35) and networks per analysis (25), humans as the selected specie, all tissues and primary cells. The resulting networks were scored based on the fold change provided by Cuffdiff as log2 (fold change) for each gene. The obtained p-values correspond to the Fisher’s exact test, with the null hypothesis that the molecules within the networks are connected based on chance.
Additional Information
How to cite this article: Córdoba, S. et al. Endometrial gene expression profile of pregnant sows with extreme phenotypes for reproductive efficiency. Sci. Rep. 5, 14416; doi: 10.1038/srep14416 (2015).
References
Rydhmer, L. Genetics of sow reproduction, including puberty, oestrus, pregnancy, farrowing and lactation. Livest. Prod. Sci. 66, 1–12 (2000).
Spötter, A. & Distl, O. Genetic approaches to the improvement of fertility traits in the pig. Vet. J. 172, 234–47 (2006).
Pope, W. F. & First, N. L. Factors affecting the survival of pig embryos. Theriogenology 23, 91–105 (1985).
Gu, T. et al. Endometrial gene expression profiling in pregnant Meishan and Yorkshire pigs on day 12 of gestation. BMC Genomics 15, 156 (2014).
Haley, C. S. & Lee, G. J. Genetic basis of prolificacy in Meishan pigs. J. Reprod. Fertil. Suppl. 48, 247–59 (1993).
Noguera, J. L. et al. A bi-dimensional genome scan for prolificacy traits in pigs shows the existence of multiple epistatic QTL. BMC Genomics 10, 636 (2009).
Rothschild, M. F., Hu, Z. & Jiang, Z. Advances in QTL mapping in pigs. Int. J. Biol. Sci. 3, 192–7 (2007).
Rathje, T. A., Rohrer, G. A. & Johnson, R. K. Evidence for quantitative trait loci affecting ovulation rate in pigs. J. Anim. Sci. 75, 1486–94 (1997).
Rohrer, G. A. et al. A comprehensive map of the porcine genome. Genome Res. 6, 371–91 (1996).
Wilkie, P. J. et al. A genomic scan of porcine reproductive traits reveals possible quantitative trait loci (QTLs) for number of corpora lutea. Mamm. Genome 10, 573–8 (1999).
Campbell, E. M. G., Nonneman, D. & Rohrer, G. A. Fine mapping a quantitative trait locus affecting ovulation rate in swine on chromosome 8. J. Anim. Sci. 81, 1706–14 (2003).
De Koning, D. J. et al. Detection and characterization of quantitative trait loci for meat quality traits in pigs. J. Anim. Sci. 79, 2812–9 (2001).
King, A. H., Jiang, Z., Gibson, J. P., Haley, C. S. & Archibald, A. L. Mapping quantitative trait loci affecting female reproductive traits on porcine chromosome 8. Biol. Reprod. 68, 2172–9 (2003).
Cassady, J. P. et al. Identification of quantitative trait loci affecting reproduction in pigs. J. Anim. Sci. 79, 623–33 (2001).
Rohrer, G. A., Ford, J. J., Wise, T. H., Vallet, J. L. & Christenson, R. K. Identification of quantitative trait loci affecting female reproductive traits in a multigeneration Meishan-White composite swine population. J. Anim. Sci. 77, 1385–91 (1999).
Rothschild, M. F. Genetics and reproduction in the pig. Anim. Reprod. Sci. 42, 143–151 (1996).
Samborski, A., Graf, A., Krebs, S., Kessler, B. & Bauersachs, S. Deep sequencing of the porcine endometrial transcriptome on day 14 of pregnancy. Biol. Reprod. 88, 84 (2013).
Bauersachs, S. & Wolf, E. Transcriptome analyses of bovine, porcine and equine endometrium during the pre-implantation phase. Anim. Reprod. Sci. 134, 84–94 (2012).
Franczak, A., Wojciechowicz, B. & Kotwica, G. Transcriptomic analysis of the porcine endometrium during early pregnancy and the estrous cycle. Reprod. Biol. 13, 229–37 (2013).
Liu, X., Xu, Z., Zhu, L., Liao, S. & Guo, W. Transcriptome Analysis of Porcine Thymus following Porcine Cytomegalovirus Infection. PLoS One 9, e113921 (2014).
Siqueira, F. M. et al. Unravelling the transcriptome profile of the Swine respiratory tract mycoplasmas. PLoS One 9, e110327 (2014).
Ramayo-Caldas, Y. et al. Liver transcriptome profile in pigs with extreme phenotypes of intramuscular fatty acid composition. BMC Genomics 13, 547 (2012).
Corominas, J. et al. Analysis of porcine adipose tissue transcriptome reveals differences in de novo fatty acid synthesis in pigs with divergent muscle fatty acid composition. BMC Genomics 14, 843 (2013).
Puig-Oliveras, A. et al. Differences in muscle transcriptome among pigs phenotypically extreme for fatty acid composition. PLoS One 9, e99720 (2014).
Gu, T. et al. Endometrial gene expression profiling in pregnant Meishan and Yorkshire pigs on day 12 of gestation. BMC Genomics 15, 156 (2014).
Zhang, H. et al. Differential gene expression in the endometrium on gestation day 12 provides insight into sow prolificacy. BMC Genomics 14, 45 (2013).
Zhou, Q.-Y. et al. Detection of differentially expressed genes between Erhualian and Large White placentas on day 75 and 90 of gestation. BMC Genomics 10, 337 (2009).
Kotlabova, K., Doucha, J. & Hromadnikova, I. Placental-specific microRNA in maternal circulation–identification of appropriate pregnancy-associated microRNAs with diagnostic potential. J. Reprod. Immunol. 89, 185–91 (2011).
Tang, F. et al. Maternal microRNAs are essential for mouse zygotic development. Genes Dev. 21, 644–8 (2007).
Yu, Z., Jian, Z., Shen, S.-H., Purisima, E. & Wang, E. Global analysis of microRNA target gene expression reveals that miRNA targets are lower expressed in mature mouse and Drosophila tissues than in the embryos. Nucleic Acids Res. 35, 152–64 (2007).
Bidanel, J. Estimation of crossbreeding parameters between Large White and Meishan porcine breeds. III. Dominance and epistatic components of heterosis on reproductive traits. Genet. Sel. Evol. 25, 263 (1993).
Silió L., R. J. & T. M. A. in Porc. ibérico Asp. claves ( Buxadé, C. & Daza, A. ) 125–149 (S.A. Mundi-Prensa Libros, 2001).
Vallet, J. L. & Freking, B. A. Differences in placental structure during gestation associated with large and small pig fetuses. J. Anim. Sci. 85, 3267–75 (2007).
Ford, S. P., Vonnahme, K. A. & Wilson, M. E. Uterine capacity in the pig reflects a combination of uterine environment and conceptus genotype effects. J. Anim. Sci. 80, E66–E73 (2001).
Bazer, F. W. et al. Novel pathways for implantation and establishment and maintenance of pregnancy in mammals. Mol. Hum. Reprod. 16, 135–52 (2010).
Cha, J., Sun, X. & Dey, S. K. Mechanisms of implantation: strategies for successful pregnancy. Nat. Med. 18, 1754–67 (2012).
Chen, X. et al. Differential Gene Expression in Uterine Endometrium During Implantation in Pigs. Biol. Reprod. 92, 52 (2014).
Martínez-Giner, M. et al. Expression study on the porcine PTHLH gene and its relationship with sow teat number. J. Anim. Breed. Genet. 128, 344–53 (2011).
Mousa, A. a et al. Preeclampsia is associated with alterations in DNA methylation of genes involved in collagen metabolism. Am. J. Pathol. 181, 1455–63 (2012).
Fan, Y.-F., Hou, Z.-C., Yi, G.-Q., Xu, G.-Y. & Yang, N. The sodium channel gene family is specifically expressed in hen uterus and associated with eggshell quality traits. BMC Genet. 14, 90 (2013).
Ashworth, M. D. et al. Expression of porcine endometrial prostaglandin synthase during the estrous cycle and early pregnancy and following endocrine disruption of pregnancy. Biol. Reprod. 74, 1007–15 (2006).
Blitek, A., Morawska, E., Kiewisz, J. & Ziecik, A. J. Effect of conceptus secretions on HOXA10 and PTGS2 gene expression and PGE2 release in co-cultured luminal epithelial and stromal cells of the porcine endometrium at the time of early implantation. Theriogenology 76, 954–66 (2011).
Blitek, A. et al. Expression of cyclooxygenase-1 and -2 in the porcine endometrium during the oestrous cycle and early pregnancy. Reprod. Domest. Anim. 41, 251–7 (2006).
Spencer, T. E., Burghardt, R. C., Johnson, G. A. & Bazer, F. W. Conceptus signals for establishment and maintenance of pregnancy. Anim. Reprod. Sci. 82-83, 537–50 (2004).
Dey, S. K. et al. Molecular cues to implantation. Endocr. Rev. 25, 341–73 (2004).
Zhang, S. et al. Deciphering the molecular basis of uterine receptivity. Mol. Reprod. Dev. 80, 8–21 (2013).
Parisi, S. & Russo, T. Regulatory role of Klf5 in early mouse development and in embryonic stem cells. Vitam. Horm. 87, 381–97 (2011).
Sun, X. et al. Kruppel-like factor 5 (KLF5) is critical for conferring uterine receptivity to implantation. Proc. Natl. Acad. Sci. USA 109, 1145–50 (2012).
Lin, S.-C. J., Wani, M. A., Whitsett, J. A. & Wells, J. M. Klf5 regulates lineage formation in the pre-implantation mouse embryo. Development 137, 3953–63 (2010).
Wu, D. et al. Molecular characterization and identification of the E2/P4 response element in the porcine HOXA10 gene. Mol. Cell. Biochem. 374, 213–22 (2013).
Schäfers, M., Schober, O. & Hermann, S. Matrix-metalloproteinases as imaging targets for inflammatory activity in atherosclerotic plaques. J. Nucl. Med. 51, 663–6 (2010).
Wang, H. et al. Functionally significant SNP MMP8 promoter haplotypes and preterm premature rupture of membranes (PPROM). Hum. Mol. Genet. 13, 2659–69 (2004).
Friedman, J. R. & Kaestner, K. H. The Foxa family of transcription factors in development and metabolism. Cell. Mol. Life Sci. 63, 2317–28 (2006).
Chakrabarty, A. et al. MicroRNA regulation of cyclooxygenase-2 during embryo implantation. Proc. Natl. Acad. Sci. USA 104, 15144–9 (2007).
Penzkofer, D. et al. Phenotypic characterization of miR-92a-/- mice reveals an important function of miR-92a in skeletal development. PLoS One 9, e101153 (2014).
Hawkins, S. M. et al. Dysregulation of uterine signaling pathways in progesterone receptor-Cre knockout of dicer. Mol. Endocrinol. 26, 1552–66 (2012).
Kowalewska, M. et al. microRNAs in uterine sarcomas and mixed epithelial-mesenchymal uterine tumors: a preliminary report. Tumour Biol. 34, 2153–60 (2013).
Bonauer, A. & Dimmeler, S. The microRNA-17-92 cluster: still a miRacle? Cell Cycle 8, 3866–73 (2009).
Bonauer, A. et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324, 1710–3 (2009).
Su, L., Zhao, S., Zhu, M. & Yu, M. Differential expression of microRNAs in porcine placentas on days 30 and 90 of gestation. Reprod. Fertil. Dev. 22, 1175–82 (2010).
Balcells Ortega, I. Deciphering the genetic architecture of prolificacy related traits in an experimental Iberian x Meishan F2 intercross. PhD dissertation, Universitat Autònoma de Barcelona, Department de Ciència Animal i dels Aliments, May (2012).
Calin, G. A. & Croce, C. M. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 66, 7390–4 (2006).
Dominguez, F., Pellicer, A. & Simon, C. The chemokine connection: hormonal and embryonic regulation at the human maternal-embryonic interface–a review. Placenta 24 Suppl B, S48–55 (2003).
Simón, C., Caballero-Campo, P., García-Velasco, J. A. & Pellicer, A. Potential implications of chemokines in reproductive function: an attractive idea. J. Reprod. Immunol. 38, 169–93 (1998).
Dunne, A. & O’Neill, L. A. J. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci. STKE 2003, re3 (2003).
Rossi, M. et al. Identification of genes regulated by interleukin-1beta in human endometrial stromal cells. Reproduction 130, 721–9 (2005).
Healy, L. L., Cronin, J. G. & Sheldon, I. M. Endometrial cells sense and react to tissue damage during infection of the bovine endometrium via interleukin 1. Sci. Rep. 4, 7060 (2014).
Paradowska, E., Blach-Olszewska, Z. & Gejdel, E. Constitutive and induced cytokine production by human placenta and amniotic membrane at term. Placenta 18, 441–6 (1997).
Szarka, A., Rigó, J., Lázár, L., Beko, G. & Molvarec, A. Circulating cytokines, chemokines and adhesion molecules in normal pregnancy and preeclampsia determined by multiplex suspension array. BMC Immunol. 11, 59 (2010).
Khan, D. A., Ansari, W. M. & Khan, F. A. Pro/Anti-Inflammatory Cytokines in the Pathogenesis of Premature Coronary Artery Disease. J. Interferon Cytokine Res. 31, 561–567 (2011).
Haider, S. & Knöfler, M. Human tumour necrosis factor: physiological and pathological roles in placenta and endometrium. Placenta 30, 111–23 (2009).
Hecht, J. L. et al. Relationship Between Neonatal Blood Protein Concentrations and Placenta Histologic Characteristics in Extremely Low GA Newborns. Pediatr. Res. 69, 68–73 (2011).
Hughes, P. E. & Varley, M. A. Reproduction in the pig. (Butterworth-Heinemann Ltd, 1980).
Friedländer, M. R. et al. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 26, 407–15 (2008).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010).
Hu, Z.-L., Park, C. A., Wu, X.-L. & Reecy, J. M. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 41, D871–9 (2013).
Balcells, I., Cirera, S. & Busk, P. K. Specific and sensitive quantitative RT-PCR of miRNAs with DNA primers. BMC Biotechnol. 11, 70 (2011).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–8 (2001).
Ferraz, A. L. J. et al. Transcriptome architecture across tissues in the pig. BMC Genomics 9, 173 (2008).
Martínez-Giner, M., Noguera, J. L., Balcells, I., Fernández-Rodríguez, A. & Pena, R. N. Selection of internal control genes for real-time quantitative PCR in ovary and uterus of sows across pregnancy. PLoS One 8, e66023 (2013).
Timoneda, O., Balcells, I., Córdoba, S., Castelló, A. & Sánchez, A. Determination of reference microRNAs for relative quantification in porcine tissues. PLoS One 7, e44413 (2012).
Acknowledgements
This research was funded by the projects AGL2004–08368-C03, AGL2010-22358-C02-01 and by the Consolider-Ingenio 2010 Programme (CSD2007-00036) all from the Spanish Ministry of Economy and Competitiveness. The authors are also indebted to INRA (France) and the CIA El Dehesón del Encinar (Spain) for providing the purebred Meishan sows and Iberian boars, respectively. Funding: This research was funded by the Science and Innovation Ministry of the Spanish Government (projects AGL2010-22358-C02-01 and AGL2004–08368-C03) and also by the Consolider-Ingenio 2010 Programme (CSD2007-00036). SC is a recipient of an FPI PhD fellowship from the Science and Innovation Ministry of the Spanish Government. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Experiments conceived and designed by A.C., A.S. and S.C. Experimental material collection and F2 management: A.S., C.O. and J.N. Experiments performed by S.C. and A.C. Data analysed by S.C. and A.C. Statistical analysis performed by S.C and A.C. Paper written by S.C. Critical revisions of manuscript: A.C., A.S., C.O., I.B., J.N. and O.T. Funding obtained by AS. Research overview: A.S.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Córdoba, S., Balcells, I., Castelló, A. et al. Endometrial gene expression profile of pregnant sows with extreme phenotypes for reproductive efficiency. Sci Rep 5, 14416 (2015). https://doi.org/10.1038/srep14416
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep14416
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
