
Abstract
Human ether-à-go-go related gene (hERG) 1 channels conduct the rapid delayed rectifier K+ current (IKr) and are essential for the repolarization of the cardiac action potential. hERG1 inhibition by structurally diverse drugs may lead to life threatening arrhythmia. Putative binding determinants of hERG1 channel blockers include T623, S624 and V625 on the pore helix and residues G648, Y652 and F656, located on segment S6. We and others have previously hypothesized that additional binding determinants may be located on helix S5, which is in close contact with the S6 segments. In order to test this hypothesis, we performed a detailed investigation combining ionic current measurements with two-microelectrode voltage clamp and molecular modeling techniques. We identified a novel aromatic high affinity binding determinant for blockers located in helix S5, F557, which is equally potent as Y652. Modeling supports a direct interaction with the outer pore helix.
Similar content being viewed by others

Introduction
Human ether-à-go-go related gene (hERG) 1 channels conduct the rapid delayed rectifier K+ current (IKr) and are essential for regulating the duration of the plateau phase of the cardiac action potential1,2. Inherited loss-of-function mutations in hERG1 can lead to life threatening torsades de pointes (TdP) arrhythmia3, while gain-of-function mutations are associated with short QT syndrome4. Most frequently, TdP arrhythmia is an acquired disorder, resulting from “off-target” block of this channel by structurally diverse drugs including antiarrhythmics, antihistamines, antipsychotics and antibiotics5. Since this inhibition can lead to sudden cardiac death, several pharmaceuticals such as cisapride or terfenadine were withdrawn from the market, or had their use severely restricted6,7. Recently, the Cardiac Safety Research Consortium (CSRC) and the Food and Drug Administration (FDA) proposed a new cardiac safety paradigm labelled “Comprehensive In Vitro Proarrhythmia Assay” (CiPA). The new CiPA guidelines emphasize the importance of studying pharmacological effects of drugs on three different ion channel types including hERG, Nav1.5 and Cav1.2, which proposed to play an important role in shaping the ventricular action potential8. hERG1 blockers might also have beneficial therapeutic potential. During routine preclinical screening for hERG1, new modulators, so-called activators, have been identified. These modulators may have the potential of shortening the action potential duration9. Thus, they might be beneficial for the treatment of inherited long QT syndrome.
Great efforts have been directed toward a better understanding of the molecular and structural mechanisms of hERG1 channel drug interactions, including in vivo, in vitro and in silico approaches (for a recent reviews see Durdagi, S. et al.10 and Vandenberg, J. et al.11). Substantial progress has been made by identifying amino acids essential for drug block. The majority of hERG inhibitors are interacting with the pore module. This homo tetrameric module consists of an outer S5 helix, a pore helix P1 and an inner helix S6. They include T623, S624 and V625 from the P1 and residues G648, Y652 and F656 located on the helix S612,13,14,15. We and other groups have presented in silico modeling studies to provide qualitative16,17,18,19 and in some cases quantitative20,21 insights into drug-channel interactions. However, no consensus about the binding mode(s) has been achieved. It was previously suggested that helix S5, which is in close contact with the S6 segments, might influence drug binding17,18,22. In particular, hydrophobic binding pockets, involving the interface between two subunits and the outer S5 helix, have been discovered as binding site for some small molecule hERG activators23,24.
According to our validated homology model (hERG-KvAP-m6)22, F557 on S5 is in close contact with the side chain of Y652 on S6. Thus, we assumed it might be possible for blockers to access lateral side openings, as has been shown recently for sodium channel blockers25. In order to test this hypothesis, we combined mutagenesis, voltage clamp analysis and molecular modeling techniques. Thereby, we identified a novel aromatic high affinity binding determinant for hERG blockers in helix S5.
Results
Effect of activator binding determinant F557 on pore blockers
To determine whether phenylalanine in position 557 (see Fig. 1 for location) affects hERG inhibition, we introduced the point mutation F557L. This mutation exhibits normal P-type inactivation, as reported previously23. Both, the F557L mutant and the wild type hERG (WT) channels were expressed in Xenopus laevis oocytes and potassium currents were measured with the two-microelectrode voltage clamp technique. Six “gold standard” hERG blockers (dofetilide, haloperidol, cisapride, astemizole, amiodarone and terfenadine) were used to validate the potential impact of F557. For hERG measurements, the oocytes were clamped at a holding potential of −100 mV and depolarized to +20 mV allowing activation and inactivation. Subsequent recovery from inactivation during a repolarisation to −50 mV induced large tail currents (Fig. 2A). Channel block was estimated from tail current inhibition. Substantial shifts of the concentration response curves of F557L to the right and incomplete hERG inhibition at high concentrations compared to WT (Fig. 2B–G) illustrates strong impairment of channel block. In order to estimate the severity of this effect, we compared concentration response relationships of F557L with mutant Y652A, which is known to efficiently diminish hERG inhibition12,15. Both mutations induced comparable shifts of the concentration response curves compared to WT channels for all studied blockers. This suggests that F557L on segment S5 is an equally strong molecular determinant of hERG inhibition as the well-established putative binding determinant Y652 (Fig. 2).

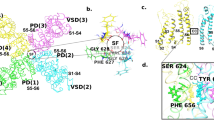
Location of tested residues and known binding determinants T623, S624, Y652 and F656.
All residues are represented as sticks in one subunit of the hERGKvAP-m6 homology model.

Mutation of F557L alters the sensitivity of hERG channel to drug block.
(A) Representative current traces of hERG and the indicated mutants in response to the pulse protocol illustrated. (B–G) Concentration-response relationships for block of WT, mutants F557L and Y652A channels by dofetilide (B) haloperidol (C) cisapride (D) astemizole (E) amiodarone (F) and terfenadine (G).
For further validation of the impact of residue F557, we estimated the inhibition of currents through WT, F557L and Y652A at concentrations 10 times the WT IC50 (=half maximal inhibitory concentration). The corresponding concentrations were 25 μM for dofetilide, 10 μM for haloperidol, 10 μM for cisapride, 3 μM for astemizole, 30 μM for amiodarone and 10 μM for terfenadine. The effects of F557L and Y652A on tail current inhibition by these drug concentrations are illustrated in Supplementary Fig. S1. Channel inhibition in F557L and Y652A by dofetilide, haloperidol, amiodarone and terfenadine was comparable. Parallel IC50 shifts to the right (>50 fold increase) of F557L and Y652A were observed in case of dofetilide. For haloperidol, we observed a 23 fold increase in IC50 relative to WT with F557L compared to a 31 fold increase in IC50 induced by Y652A. The least pronounced (minimum fold) increase in IC50 was observed for amiodarone, with 3 fold (F557L) and 5 fold (Y652A) increases, respectively. Similar parallel IC50 shift to right was observed for terfenadine with 12 fold (F557L) and 8 fold (Y652A) increase relative to WT. In case of cisapride, a 9 fold increase in IC50 was observed with F557L compared to 74 fold increase in IC50 with Y652A. Similarly, astemizole showed 6 fold shift in IC50 for F557L and 18 fold shift for Y652A. Both cisapride and astemizole showed less pronounced increase in IC50 on mutant F557L in comparison to fold increase in IC50 by Y652A (See Table 1 for all studied hERG inhibitors). Taken together, F557L shifted the concentration response curves for all studied hERG blockers to the right, ranging from a 4 fold (amiodarone) to more than 50 fold (dofetilide) increase in IC50.
Role of residues surrounding F557 on hERG inhibition
F557 was previously identified as potential interaction site for hERG agonists23,24. It was therefore interesting to investigate if other high affinity agonist binding determinants, located in the vicinity of F557, would also affect channel inhibition. Consequently, we tested residues M554 located on S5, F619 and L622 located on P1, I642 and L646 located on S6, previously identified by Garg et al.23 (for location of tested residues see Fig. 1). Figure 3A–D illustrates the effects of these mutants on current inhibition by dofetilide, haloperidol and cisapride at concentration 10 times the WT IC50. Figure 3A,B shows that the effect of dofetilide on current inhibition was not altered by mutations L622C, M554A, L646E and I642C. However, a moderate effect on channel inhibition by dofetilide was observed for F619A. As illustrated in Fig. 3C,D none of these mutations significantly affected the channel inhibition by haloperidol and cisapride, respectively.

Effects of mutations surrounding F557 on channel inhibition by dofetilide, haloperidol and cisapride.
(A) Representative currents through the indicated hERG channel mutants in control (black traces) and after steady state block by 25 μM dofetelide was reached (red traces, same voltage protocol as shown in top panel. (B–D) Inhibition of currents through the indicated mutants by dofetilide (25 μM), haloperidol (10 μM) and cisapride (9 μM). Data are represented throughout the figure as mean ± SEM. The numbers of experiments (n) are indicated as small insets within the bars. ***P < 0.001, **P < 0.01, one-way ANOVA. The value of 1 indicates no detectable decrease in current by drugs.
Choice of template determines size of lateral pore openings
To address whether residue F557 indeed influences drug block via direct π-π and/or hydrophobic interactions, or if the experimentally observed effects are allosteric, we performed in silico modeling studies. Due to the absence of hERG crystal structures, homology modeling techniques were employed to investigate drug binding. Crystal structures of several K+ channels have been determined in closed, open and putatively open inactivated states, enabling modeling of hERG in different channel states. Since all x-ray structure templates have low sequence identity to hERG, the correct choice of template/s and alignment is not trivial22. In order to minimize shortcomings based on template choice or channel state, we chose to systematically investigate the role of F557 using five different hERG homology models, based on open and/or open inactivated K+ channel templates, using our previously published alignments22. As can be seen in Supplementary Fig. S2, the choice of K+ channel template has a profound influence on the spatial arrangement of the molecular determinants Y652, F656 and F557.
In a first step, we analyzed the size of the lateral openings in the different hERG homology models using the pymol plugin caver 3.0.126. The center of residue G648 in helix S6 of all 4 chains was used as a starting point. The diameter of the narrowest “constriction” in the fenestration pathway of the different hERG models can be found in Table 2. As shown in Supplementary Fig. S3, the size of the lateral pore openings varies, depending on the choice of template. Lateral pore openings towards S5, which are presumably large enough for drug interactions, are found in three models: the open state model based on hERGKvAP-m622, the KcsA-inactivation based model hERGKcsA-I27,28 and the MthK based model hERGMthK-L29. Consequently, only in these three models possible direct drug interactions with F557 in helix S5 were investigated; docking in all other models did not lead to any acceptable poses, despite allowing free rotation of all binding aromatics (data not shown).
Modeling suggests direct interaction of high affinity hERG blockers with F557
To investigate, if π-π or cation-π interactions between high affinity blockers and residue F557 are possible, we performed docking simulations. Figure 4 summarizes the best docking poses (highest binding affinity from Chemscore, summarized in Supplementary Table S1) obtained for each compound. In case of amiodarone, astemizole and dofetilide, the highest score poses were obtained with model hERGKvAP-m6. For cisapride, haloperidol and terfenadine, the highest ranked poses were obtained in hERGKcsA-I.

Docking poses revealing aromatic interactions with residue F557.
Left side: side view of two neighboring subunits colored in yellow and blue. Right side: top view of hERG blockers and interacting residues, presented as sticks. π-π interactions between aromatic rings are indicated by blue dots (cutoff distances: sandwich-shaped = 5Å; t-shaped = 6.5 Å; parallel displaced = 5.5 Å)56.
Generally, direct π-π interactions between drugs and the aromatic side chain of F557 are possible for all drugs. These direct interactions influence the positioning of the blockers in the central cavity. Binding poses enabling π-π interactions with F557 are characterized by protrusion into the lateral openings between two neighboring subunits, below the selectivity filter. For all studied drugs, extensive ring stacking or hydrophobic interactions with several aromatic side chains including Y652, F656 and F557 were observed. In case of haloperidol, terfenadine, cisapride and dofetilide, polar interactions with T623 and/or S624 are predicted. Further, the polarizable nitrogen of the investigated blockers in cisapride and terfenadine is positioned right below the pore helix, suggesting that the helix dipole charges contribute to binding (see Fig. 4).
Discussion
It is widely accepted that block of hERG K+ channels by structurally diverse molecules is mediated by two aromatic side chains Y652 and F65612. Mutation of either residue to alanine dramatically reduces drug potency. Modeling studies suggest direct interaction of drugs with these residues16,18,30. According to our recent hERG model22, residue F557 in helix S5 is in close proximity with Y652 (S6). This residue was identified as crucial molecular determinant for the effect of hERG activator ICA-10557423. In order to test the hypothesis that determinants of hERG activators might interact with hERG blockers, we mutated F557 and analysed the inhibition of potassium currents through mutant F557L by six “gold standard” hERG blockers dofetilide, haloperidol, terfenadine, astemizole, cisapride and amiodarone. Our study reveals that mutation F557L dramatically decreases current inhibition. To our surprise, F557L reduced the drug potency to the same extent as the well-studied mutation Y652A (Fig. 2). We further investigated if other residues in close proximity of F557 (see Fig. 1), such as F619, L622, M554, L646 and I642 also influence channel inhibition. However, except for F619A, which had a moderate effect on channel inhibition by dofetilide, none of the studied mutations significantly affected channel inhibition by the tested drugs (Fig. 3).
To investigate the role of F557 in more detail, we performed modeling studies to elucidate potential interactions between F557 in helix S5 and high affinity blockers tested in our study. Docking studies with different open or open/inactivated hERG models (Supplementary Figs S2 and S3) suggest that all studied blockers bind in the cavity, below the selectivity filter, partially protruding into lateral, hydrophobic pore openings (Fig. 4). These openings, also known as fenestrations, have first been identified in bacterial sodium channels31,32. Interestingly, the novel binding modes for hERG blockers proposed in this study resembles recently reported binding sites of brominated sodium channel blockers in the bacterial sodium channel NavMs crystal structure25. A recent x-ray analysis with quaternary ammonium blockers in KcsA, suggests that fenestrations in this potassium channel are only accessible, if rotameric changes of the aromatic side chain of residue F103 occur33.
Interestingly, reshaping of fenestrations has been suggested to play an important role in slow inactivation in bacterial sodium channels32. Thus, it is conceivable that binding of small molecules to the hydrophobic side openings affects the affinity of drugs to the inactivated state. This is further supported by recent studies, revealing the binding site for a small molecule activator in hERG at this side, which strongly attenuates inactivation23. The mechanisms, how inactivation influences drug affinity in different ion channels, is still poorly understood. Based on our modeling with different K+ channel structure templates, one might speculate that the geometry of the hydrophobic side pockets depends on the geometry of the helices and the rotameric state(s) of the aromatic side chains, which are linked to the channel state. Interestingly, previous modeling studies on hERG observed transiently occurring hydrophobic openings between subunits, exposing the central cavity to the hydrophobic core of the lipid membrane17,18. Further, x-ray and MD studies on KcsA clearly revealed a structural link between the rotameric state of F103 (located at the entrance of the fenestration) in KcsA, the intracellular gate and the selectivity filter27,34.
Taken together, all these studies suggest that binding of drugs to hydrophobic side pockets might be quite common for cation channels and simultaneously, may play a role in inactivation gating. However, this issue clearly warrants further research.
The novel binding mode for high affinity blockers proposed in this study, is in good agreement with most published experimental data available for hERG blockers12,14,15,35,36,37,38,39,40,41. However, tandem dimer mutant studies30 for cisapride and terfenadine reporting on the number of aromatic interactions are not fully consistent with the proposed binding mode for these drugs from our study. In particular, the arrangement of Y652 and F656 from one subunit and the Y652 side chain from an adjacent subunit are not seen in any poses, where F557 directly interacts with the drug. Such poses are only possible in the conventional binding mode. A plausible explanation for this discrepancy is the existence of several binding modes for hERG blockers. This would also explain, why so many different hERG binding modes have been predicted in numerous previous studies16,18,21,42,43,44,45. The heterogeneous nature of drug binding in hERG might further explain why so many structurally diverse compounds can block this channel. Additionally, it is more and more appreciated that small molecule interactions with ion channels can occur at many sites46,47,48. Alternatively, we cannot exclude that F557 does not contribute directly to drug binding but, instead, modulates channel block allosterically. However, our simulation studies strongly support a direct interaction.
Summarizing, our study reveals a putative novel high affinity binding determinant for hERG blockers. Further, we propose the existence of a novel hydrophobic binding site, at the fenestrations, additionally to the conventional binding site in the central cavity.
Materials and Methods
Oocyte electrophysiology
cDNAs of hERG (accession number NP000229) and constructs F557L, F619A, L622C, M554A, L646E, I642C were kindly provided by MC Sanguinetti (University of Utah, UT, USA) and JI Vandenberg (Molecular Cardiology and Biophysics Division, Victor Chang Cardiac Research Institute, Darlinghurst, New South Wales). Synthesis of capped runoff complementary ribonucleic acid (cRNA) transcripts from linearized complementary deoxyribonucleic acid (cDNA) templates and injection of cRNA were performed as described previously49. Oocytes from the South African clawed frog, Xenopus laevis, (NASCO, Fort Atkinson, WI, USA) were prepared as follows: After 15 min exposure of female Xenopus laevis to the anaesthetic (0.2% solution of MS-222; the methane sulfonate salt of 3-aminobenzoic acid ethyl ester; Sigma), parts of the ovary tissue were surgically removed. Oocytes were defolliculated by enzymatical treatment with 2 mg/mL collagenase type 1 A (Sigma) and further by mechanical removal of follicular layer using forceps. Stage V–VI oocytes were selected and injected with the WT hERG-encoding cRNA. Injected oocytes were stored at 18 °C in ND96 bath solution (96 mM sodium chloride, 2 mM potassium chloride, 1 mM magnesium chloride, 1.8 mM calcium chloride, 5 mM HEPES; pH 7.4, titrated with NaOH) containing 1% penicillin-streptomycin solution. All chemicals used were purchased from Sigma- Aldrich Chemie GmbH, Taufkirchen, Germany.
Currents through hERG channels were studied 1–2 days after microinjection of the cRNA using the two-microelectrode voltage clamp technique. ND96 was used as an extracellular recording solution. Voltage-recording and current-injecting microelectrodes were filled with 3 M KCl and had resistances between 0.3 and 2 MΩ. Endogenous currents (estimated in oocytes injected with water) did not exceed 0.15 μA. Currents >5 μA were discarded to minimize voltage clamp errors. A precondition for all measurements was the achievement of stable peak current amplitudes over periods of 10 min after an initial run-up period. All drugs were applied by means of semi-fast perfusion system50. Ionic currents were recorded with a Turbo Tec 03X Amplifier (npi electronic, GmbH, Tamm, Germany) and digitized with a Digidata 1322A digitizer (Axon Instruments Inc., Union City, CA, USA). The pClamp software package version 9.2 (Axon Instruments Inc.) was used for data acquisition. Microcal Origin 7.0 was employed for analysis and curve fitting. Currents through wild type hERG channels and channel mutants were studied at a holding potential of −100 mV using a 2 step voltage protocol. A conditioning 300 ms depolarizing step to +20 mV was followed by a 300 ms repolarization to −50 mV. Repolarisation induced large tail currents. Drug effects were analyzed after a 15–20 minute “run-up” phase was completed. Drug effects were estimated from peak tail current inhibition at −50 mV after steady state was reached. Tail current concentration–inhibition curves were fitted using the Hill equation (see equation (1)).

In the hill equation, IC50 is the concentration at which hERG inhibition is half-maximal, C is the applied drug concentration in μM, A is the fraction of hERG current that is not blocked and nH is the Hill coefficient. Data points represent means ± s.e. from at least three oocytes from >2 batches; Statistical significance was calculated using a one-way ANOVA followed by a bonferroni’s multiple comparison test **p < 0.01 and ***p < 0.001, respectively. All the studied compounds were purchased from Sigma Aldrich. All compounds were first dissolved in dimethyl sulphoxide (DMSO) to prepare a 10 mM stock solution and stored at −20 °C. Drug stocks were diluted to the required concentration in extracellular solution on the day of each experiment. The maximal DMSO concentration in the bath (0.1%) did not affect hERG currents.
Ethical statement
All experiments involving animals were approved by the Austrian Animal Experimentation Ethics Board in accordance with the European convention for the protection of vertebrate animals used for experimental and other scientific purposes ETS no. 123, which is in line with the EU Directive 2010/63/EU (GZ 66.006/0019-C/GT/2007). The methods were carried out in accordance with the approved guidelines.
Homology modeling
Homology models of the open and inactivated hERG pore domain, based on different K+ channel templates, were generated with Modeller 9v12. Templates include the pore domain of the KcsA crystal structure (Protein Data Bank (pdb) ID 3F5W27,51), the Kv1.2 structure (pdb ID: 3LUT52) and the MthK x-ray structures (pdb ID: 1LNQ29 and 3LDD53). Modeling details, including coordinates for the KvAP based open conformation, have been described previously22 (model m6). Alignments of the models are described in Stary et al.22 (see Fig. 1 in ref. 22).
Analysis of fenestrations
Caver parameters were used as previously described by Kaczmarski & Corry54 (Probe_radius = 0.8 Å; Shell_radius = 15 Å; Shell_depth = 15 Å; Clustering_threshold = 10 Å; Number_of_approximating_balls = 12). The starting point was selected using the center of the G648 residue in the S6 of all 4 chains.
Drug docking and parametrization
Docking was performed using the program Gold 4.0.1 and the implemented Gold scoring function55. hERG homology models in the open state, based on the KvAP template termed “hERG-KvAP-m6”22, a high resolution MthK structure in open conformation (3LDD53) termed “hERG-MthK-O”, the high resolution structure of Kv1.2 (3LUT52), termed “hERG-Kv1.2” and two putatively inactivated state models based on the KcsA template 3F5W27,51, termed “hERG-KcsA-I” and the low resolution x-ray MthK structure 1LNQ29 termed “hERG-MthK-I” were used for docking22. The binding site in all models was defined by setting Y652 of one SU as the center and the radius to 10 Å. This binding site thereby included the reported binding determinants Y652, F656, T623, S624 and the putative interacting aromatic amino acids F557 and F619. The rotameric state of these amino acids was set to flexible, allowing free sampling of side chain conformations. For all drugs, the central nitrogen was protonated and used in their charged form. The 20 best-ranked poses of each drug docking run were visually inspected.
Additional Information
How to cite this article: Saxena, P. et al. New potential binding determinant for hERG channel inhibitors. Sci. Rep. 6, 24182; doi: 10.1038/srep24182 (2016).
References
Sanguinetti, M. C., Jiang, C. & Keating, M. T. A mechanistic link between an inherited and an acquired cardiac arrthytmia: HERG encodes the IKr potassium channel. Cell 81, 299–307 (1995).
Trudeau, M. C., Warmke, J. W., Ganetyky, B. & Robertson, G. A. HERG, a Human Inward Rectifier in the Voltage-Gated Potassium Channel Family. Science 269, 92–95 (1995).
Curran, M. E. et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 (1995).
Brugada, R. et al. Sudden Death Associated with Short-QT Syndrome Linked to Mutations in HERG. Circulation 109, 30–35 (2004).
Fermini, B. & Fossa, A. A. The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2, 439–47 (2003).
Zhou, Z., Gong, Q. & January, C. T. Blockage of the HERG human cardiac K+ channel by the gastrointestinal prokinetic agent cisapride. Rapid Commun. Am. Physiol. Soc. 273, H2534–H2538 (1997).
Roy, M. L., Dumaine, R. & Brown, A. M. HERG, a Primary Human Ventricular Target of the Nonsedating Antihistamine Terfenadine. Circulation 94, 817–823 (1996).
Kramer, J. et al. MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci. Rep. 3, 2100 (2013).
Sanguinetti, M. C. HERG1 channel agonists and cardiac arrhythmia. Curr. Opin. Pharmacol. 15, 22–27 (2014).
Durdagi, S. et al. Insights into the molecular mechanism of hERG1 channel activation and blockade by drugs. Curr. Med. Chem. 17, 3514–3532 (2010).
Vandenberg, J. I. et al. hERG K+ Channels: Structure, Function and Clinical Significance. Physiol. Rev. 92, 1393–1478 (2012).
Mitcheson, J. S., Chen, J., Lin, M., Culberson, C. & Sanguinetti, M. C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. USA 97, 12329–33 (2000).
Kamiya, K., Niwa, R., Morishima, M., Honjo, H. & Sanguinetti, M. C. Molecular Determinants of hERG Channel Block by Terfenadine and Cisapride. J. Pharmacol. Sci. 108, 301–307 (2008).
Lees-Miller, J. P., Duan, Y., Teng, G. Q. & Duff, H. J. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol. Pharmacol. 57, 367–374 (2000).
Fernandez, D., Ghanta, A., Kauffman, G. W. & Sanguinetti, M. C. Physicochemical features of the HERG channel drug binding site. J. Biol. Chem. 279, 10120–7 (2004).
Dempsey, C. E., Wright, D., Colenso, C. K., Sessions, R. B. & Hancox, J. C. Assessing hERG pore models as templates for drug docking using published experimental constraints: the inactivated state in the context of drug block. J. Chem. Inf. Model. 54, 601–12 (2014).
Schmidtke, P., Ciantar, M., Theret, I. & Ducrot, P. Dynamics of hERG Closure Allow Novel Insights into hERG Blocking by Small Molecules. J. Chem. Inf. Model. 54, 2320–2333 (2014).
Zachariae, U., Giordanetto, F. & Leach, A. G. Side chain flexibilities in the human ether-a-go-go related gene potassium channel (hERG) together with matched-pair binding studies suggest a new binding mode for channel blockers. J. Med. Chem. 52, 4266–76 (2009).
Durdagi, S., Duff, H. J. & Noskov, S. Y. Combined receptor and ligand-based approach to the universal pharmacophore model development for studies of drug blockade to the hERG1 pore domain. J. Chem. Inf. Model. 51, 463–474 (2011).
Boukharta, L., Ker, H., Stary-Weinzinger, A., Groot, B. L. De & Åqvist, J. Computer Simulations of Structure-Activity Relationships for hERG Channel Blockers. Biochemistry 50, 6146–6156 (2011).
Osterberg, F. & Aqvist, J. Exploring blocker binding to a homology model of the open hERG K+ channel using docking and molecular dynamics methods. FEBS Lett. 579, 2939–44 (2005).
Stary, A. et al. Toward a consensus model of the HERG potassium channel. ChemMedChem 5, 455–67 (2010).
Garg, V., Stary-Weinzinger, A., Sachse, F. & Sanguinetti, M. C. Molecular determinants for activation of human ether-à-go-go-related gene 1 potassium channels by 3-nitro-n-(4-phenoxyphenyl) benzamide. Mol. Pharmacol. 80, 630–7 (2011).
Perry, M., Sachse, F. B., Abbruzzese, J. & Sanguinetti, M. C. PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc. Natl. Acad. Sci. USA 106, 20075–80 (2009).
Bagnéris, C. et al. Prokaryotic NavMs channel as a structural and functional model for eukaryotic sodium channel antagonism. Proc. Natl. Acad. Sci. USA 111, 8428–33 (2014).
Chovancova, E. et al. CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 8, e1002708 (2012).
Cuello, L. G., Jogini, V., Cortes, D. M. & Perozo, E. Structural mechanism of C-type inactivation in K+ channels. Nature 466, 203–208 (2010).
Cuello, L. G. et al. Structural basis for the coupling between activation and inactivation gates in K+ channels. Nature 466, 272–275 (2010).
Jiang, Y. et al. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417, 515–22 (2002).
Imai, Y. N., Ryu, S. & Oiki, S. Docking Model of Drug Binding to the Human Ether-a-go-go Potassium Channel Guided by Tandem Dimer Mutant Patch-Clamp Data : A Synergic Approach. J. Med. Chem. 52, 1630–1638 (2009).
Payandeh, J., Scheuer, T., Zheng, N. & Catterall, W. A. The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (2011).
Payandeh, J., Gamal El-Din, T. M., Scheuer, T., Zheng, N. & Catterall, W. A. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139 (2012).
Lenaeus, M. J., Burdette, D., Wagner, T., Focia, P. J. & Gross, A. Structures of KcsA in Complex with Symmetrical Quaternary Ammonium Compounds Reveal a Hydrophobic Binding Site. Biochemistry 53, 5365–5373 (2014).
Pan, A. C., Cuello, L. G., Perozo, E. & Roux, B. Thermodynamic coupling between activation and inactivation gating in potassium channels revealed by free energy molecular dynamics simulations. J. Gen. Physiol. 138, 571–580 (2011).
Kamiya, K., Niwa, R., Mitcheson, J. S. & Sanguinetti, M. C. Molecular Determinants of hERG Channel Block. Mol. Pharmacol. 69, 1709–1716 (2006).
Witchel, H. J. et al. The Low-Potency, Voltage-Dependent HERG Blocker Propafenone - Molecular Determinants and Drug Trapping. Mol. Pharmacol. 66, 1201–1212 (2004).
Ficker, E., Jarolimek, W., Kiehn, J., Baumann, A. & Brown, A. M. Molecular determinants of dofetilide block of HERG K+ channels. Circ. Res. 82, 386–95 (1998).
Suessbrich, H. et al. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br. J. Pharmacol. 120, 968–974 (1997).
Gómez-Varela, D. et al. Different relevance of inactivation and F468 residue in the mechanisms of hEag1 channel blockage by astemizole, imipramine and dofetilide. FEBS Lett. 580, 5059–66 (2006).
Ridley, J. M., Milnes, J. T., Witchel, H. J. & Hancox, J. C. High affinity HERG K(+) channel blockade by the antiarrhythmic agent dronedarone: resistance to mutations of the S6 residues Y652 and F656. Biochem. Biophys. Res. Commun. 325, 883–91 (2004).
Ficker, E., Obejero-Paz, C. A., Zhao, S. & Brown, A. M. The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J. Biol. Chem. 277, 4989–98 (2002).
Farid, R., Day, T., Friesner, R. A. & Pearlstein, R. A. New insights about HERG blockade obtained from protein modeling, potential energy mapping and docking studies. Bioorg. Med. Chem. 14, 3160–73 (2006).
Stansfeld, P. J. et al. Drug Block of the hERG Potassium Channel : Insight From Modeling. Proteins 68, 568–580 (2007).
Durdagi, S., Deshpande, S., Duff, H. J. & Noskov, S. Y. Modeling of open, closed and open-inactivated states of the hERG1 channel: structural mechanisms of the state-dependent drug binding. J. Chem. Inf. Model. 52, 2760–74 (2012).
Köpfer, D. A. et al. A Molecular Switch Driving Inactivation in the Cardiac K+ Channel hERG. PLoS One 7, e41023–e41023 (2012).
Marzian, S. et al. Side pocket provides the basis for a new mechanism of Kv channel – specific inhibition. Nat. Chem. Biol. 9, 507–513 (2013).
Wulff, H. & Yarov-Yarovoy, V. Channels: Sticking to nooks and crannies. Nat. Chem. Biol. 9, 473–474 (2013).
Du, Y. et al. Molecular evidence for dual pyrethroid-receptor sites on a mosquito sodium channel. Proc. Natl. Acad. Sci. 110, 11785–11790 (2013).
Windisch, A. et al. Trapping and dissociation of propafenone derivatives in HERG channels. Br. J. Pharmacol. 162, 1542–52 (2011).
Baburin, I., Beyl, S. & Hering, S. Automated fast perfusion of Xenopus oocytes for drug screening. Pflügers Arch. Eur. J. Pharmacol. 453, 117–123 (2006).
Uysal, S. et al. Crystal structure of full-length KcsA in its closed conformation. Proc. Natl. Acad. Sci. USA 106, 6644–6649 (2009).
Chen, X., Wang, Q., Ni, F. & Ma, J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. 107, 11352–11357 (2010).
Ye, S., Li, Y. & Jiang, Y. Novel insights into K+ selectivity from high-resolution structures of an open K+ channel pore. Nat. Struct. Mol. Biol. 17, 1019–1023 (2010).
Kaczmarski, J. A. & Corry, B. Investigating the size and dynamics of voltage-gated sodium channel fenestrations. Channels (Austin). 8, 264–77 (2014).
Cambridge Crystallographic Data Centre (CCDC)., GOLD. (2008). http://www.ccdc.cam.ac.uk (Accessed: 11th December 2015).
Tsuzuki, S., Honda, K., Uchimaru, T., Mikami, M. & Tanabe, K. Origin of Attraction and Directionality of the π/π Interaction: Model Chemistry Calculations of Benzene Dimer Interaction. J. Am. Chem. Soc. 124, 104–112 (2001).
Acknowledgements
We kindly thank Jamie I. Vandenberg and Michael C. Sanguinetti for providing the hERG mutants. This work is supported by Austrian Science Fund (FWF; http://www.fwf.ac.at). PS, TL and EMZP are supported by the doctoral programme ‘molecular drug targets’ funded by FWF W1232. ASW is supported by the Wiener Hochschuljubiläumsfonds (grant no. H-304013/2014). SH and ASW are supported by grant FWF P22395.
Author information
Authors and Affiliations
Contributions
P.S., E.M.Z.P., T.L. and A.S.W. performed research, E.T., S.H. and A.S.W. designed the study, P.S., E.M.Z.P., T.L., A.H., A.W., E.T., S.H. and A.S.W. analysed data, P.S., E.M.Z.P., E.T., S.H. and A.S.W. wrote the paper. All authors reviewed manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Saxena, P., Zangerl-Plessl, EM., Linder, T. et al. New potential binding determinant for hERG channel inhibitors. Sci Rep 6, 24182 (2016). https://doi.org/10.1038/srep24182
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep24182
This article is cited by
-
Inhibition of the hERG potassium channel by phenanthrene: a polycyclic aromatic hydrocarbon pollutant
Cellular and Molecular Life Sciences (2021)
-
A structure-based computational workflow to predict liability and binding modes of small molecules to hERG
Scientific Reports (2020)
-
On the potential of in vitro organ-chip models to define temporal pharmacokinetic-pharmacodynamic relationships
Scientific Reports (2019)
-
Calculation of absolute binding free energies between the hERG channel and structurally diverse drugs
Scientific Reports (2019)
-
Investigating the state dependence of drug binding in hERG channels using a trapped-open channel phenotype
Scientific Reports (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
