
Abstract
About 40–60% of patients with late-onset Alzheimer’s disease (AD) develop psychosis, which represents a distinct phenotype of more severe cognitive and functional deficits. The estimated heritability of AD+P is ~61%, which makes it a good target for genetic mapping. We performed a genome-wide copy-number variation (CNV) study on 496 AD cases with psychosis (AD+P), 639 AD subjects with intermediate psychosis (AD intermediate P) and 156 AD subjects without psychosis (AD−P) who were recruited at the University of Pittsburgh Alzheimer's Disease Research Center using over 1 million single-nucleotide polymorphisms (SNPs) and CNV markers. CNV load analysis found no significant difference in total and average CNV length and CNV number in the AD+P or AD intermediate P groups compared with the AD−P group. Our analysis revealed a marginally significant lower number of duplication events in AD+P cases compared with AD−P controls (P=0.059) using multivariable regression model. The most interesting finding was the presence of a genome-wide significant duplication in the APC2 gene on chromosome 19, which was protective against developing AD+P (odds ratio=0.42; P=7.2E−10). We also observed suggestive associations of duplications with AD+P in the SET (P=1.95E−06), JAG2 (P=5.01E−07) and ZFPM1 (P=2.13E−07) genes and marginal association of a deletion in CNTLN (P=8.87E−04). We have identified potential novel loci for psychosis in Alzheimer’s disease that warrant follow-up in large-scale independent studies.
Similar content being viewed by others

Introduction
Alzheimer's disease (AD), especially the late-onset form, is the leading cause of dementia among older persons. Twin studies have shown that genetic factors have an important role in explaining AD with up to 79% heritability.1 Beyond the well-established apolipoprotein E association, several susceptibility loci have been identified by large-scale genome-wide association studies.2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 However, single-nucleotide polymorphisms (SNPs) at all those loci explain <50% of AD heritability.
To identify additional genetic factors related to AD, genome-wide scans for copy-number variants (CNVs) have been conducted recently.16, 17, 18, 19, 20, 21, 22, 23, 24 Swaminathan et al.18 reported a trend for reduced levels of deletions and duplications in AD cases compared with controls; whereas Ghani et al.20 found no significant differences between cases and controls in CNV rate, distribution of deletions or duplications, total or average CNV size or number of genes affected by CNVs, except for a nominal association between AD and a duplication on 15q11.2. Chapman et al.22 reported a limited contribution of rare CNVs to AD risk, but proposed that healthy elderly individuals have a reduced rate of large deletions. The inconsistent genome-wide CNV findings in AD may partially be due to its heterogeneous nature.
About 40–60% of late-onset AD patients will develop psychosis. AD+P has more severe cognitive and functional deficits. It has more rapid deterioration than AD without psychosis and represents a distinct phenotype with a genetic basis. The estimated heritability of AD+P is about 61%;25 however the genetic variants that explain this high heritability are unknown. The first genome-wide association study of AD+P was performed recently by Hollingworth et al.26 They have identified several suggestive loci that were associated with AD+P when compared with AD−P and/or general population, but none of them were genome-wide significant. To systematically identify the genetic variants that account for additional heritability of AD+P, we conducted a genome-wide CNV study of AD+P using the Illumina HumanOmni1-Quad BeadChip (San Diego, CA, USA).
Materials and methods
Subjects
Subjects were participants in a study on the genetics of AD, which recruited 1440 AD cases (mean age-at-onset 72.6±6.4 years) through the University of Pittsburgh Alzheimer's Disease Research Center and 1000 older controls (mean age 74.07±6.20 years). All the participants were examined with detailed clinical, laboratory and neuroimaging evaluations.27, 28 Data from 1291 AD patients that passed stringent quality control criteria from our previous genome-wide study2 were used in this study. Subjects with AD met criteria for either probable or definite AD according to criteria set by the National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s disease and Related Disorders Association and the Consortium to Establish a Registry for Alzheimer’s disease.29 Subjects with evidence of persistent and or recurrent delusions or hallucinations, defined by the Consortium to Establish a Registry for Alzheimer’s disease behavior rating scale30 items for psychotic features (items 33–35) rated as occurring three or more times in the past month at any visit, were classified as AD+P, as previously described.31 Subjects with evidence of infrequent distortions of thought or perception, defined by any of these items rated as occurring one to two times in the past month, were classified as ‘indeterminate psychosis’ as such symptoms can result from a variety of sources. Subjects rated as no occurrence of possible psychotic symptoms at all visits were classified as AD−P. Subjects were excluded if they had a previous history of schizophrenia (SCZ) or bipolar disorder. Details on the study population characteristics can be found elsewhere.2, 26 Among the 1291 AD subjects, 496 were classified as AD+P, 639 as AD with ‘indeterminate’ psychosis and 156 as AD−P. Because the ‘indeterminate’ group is actually an ‘intermediate’ group and not an ‘unknown’ group, we include them in this study to increase statistical power.
Illumina assay for CNV discovery
All subjects were genotyped using the high-throughput genome-wide SNP Illumina HumanOmni1-Quad BeadChip (containing 1 016 423 SNPs and/or CNVs).2 We generated CNV calls using the PennCNV software (2009 Aug27 version).32 PennCNV is a Hidden Markov Model (HMM)-based method. It uses the log R ratio (LRR) and B allele frequency (BAF) measures computed from the signal intensity files by Beadstudio to detect the CNVs. The simultaneous analysis of intensity data and genotype data in the same experimental setting generated high accurate definition of normal diploid states and any deviation from norm.
CNV quality control
Details on the quality control parameters applied on the samples and markers have been described previously.2, 26 Criteria for exclusion of samples included the consent and diagnostic criteria, high genotyping failure rate and/or cryptic relatedness. Briefly, only samples with call rates >98% were retained. Samples were filtered if mean X-chromosome heterozygosity ⩾0.02 for males or outside the range of 0.25~0.4 for females. Genetic outliers and those with evidence of relatedness (IBD estimate ⩾0.4) or non-European ancestry based on genotype data were also excluded. After filtering, 496 AD+P cases, 639 AD intermediate P, 156 AD−P controls were retained. Markers were excluded if they had a genotype missing rate of >0.02. Markers were examined to determine whether missingness depended on case/control status or the genotyping batch. To call CNVs, we used the GC model wave adjustment procedure in PennCNV to smooth out genomic waving, since wave artifacts correlating with GC content and resulting from hybridization bias of low full-length DNA quantity can interfere with the accurate inference of CNVs. After GC model adjustment, we filtered the samples that met the criterion of LRR standard deviation ⩾0.3, and |GC base pair wave factor| >0.05. About 10% of all samples (122 subjects) were further removed. A total 440 AD+P cases, 593 AD intermediate P, 136 AD−P controls were used in the final analysis. Additional QC was applied at the CNV level by excluding CNV calls <5 kb in length and spanning <5 probes.33 All procedures followed the user guidelines of PennCNV. Table 1 summarizes the number of samples in different categories after quality control measures.
Statistical analysis of CNVs
Complex CNV overlap is simplified by producing SNP-based statistics. The CNV frequencies between groups were compared at each SNP using an ordered logistic regression model. Psychosis status was treated as an ordered categorical outcome: AD−P, AD with intermediate psychosis and AD+P. The predictor was CNV states. Copy number=2 per individual was considered normal. Deletion and duplication CNVs were defined as CNV calls with copy number <2 and >2, respectively. Deletion and duplication CNVs and normal copy number were coded as separate variables (that is, categorical model with three categories). To adjust for the confounding effect of population stratification, the first four principal components of population structure were included as covariates in the regression model. Age was also adjusted for.
Multiple testing was corrected using the Benjamini–Hochberg false discovery rate. We reported associations with false discovery rate <0.01. We reported statistical local minimums to narrow the association in reference to a region of nominal significance. Regions of significance ranging within a power of 10 are reported, as previously described.34 The resulting nominally significant CNV regions were excluded if they met either of the following criteria: (i) residing on telomere- or centromere-proximal cytobands; (ii) located in a 'peninsula' of common CNVs arising from variation in boundary truncation of CNV calling. Statistical comparisons were performed only for CNV events observed on autosomes, due to the complexity of analyzing the X and Y chromosomes. All data analyses were conducted in R (version 3.0.1).35 Manhattan plots were used to show the genome-wide P-values at each marker. As a final step to confirm the validity of events with significantly frequency differences, LRR and BAF plots were visually inspected to ensure that called CN gains and losses were visually evident and not simply artifacts of the calling algorithm. Human NCBI Build 36 (hg18) was used for assigning chromosome locations.
CNV load
For each individual, we also defined several measures of autosomal genomic CNV burden and compared mean values between phenotypic groups. CNV burden for each individual was defined in three distinct ways; (1) the total length of genomic DNA involved in identified CNV events; (2) average length of CNVs; and (3) total number of CNVs. Comparison of CNV burden among the groups was conducted using linear regression in R. The outcomes were CNV burdens, the predictor was three AD groups, which were coded as separate variables (that is, categorical model with three categories).
Results
Overall CNV burden
Clinical characteristics of the sample can be found in Table 1. We calculated the overall CNV burden including total CNV length, average CNV length and number of observed CNV events in each group of samples and also stratified copy number into duplications and deletions. Comparison of total and average CNV length and CNV number revealed no significant difference in the AD+P or AD intermediate P groups compared with the AD−P groups (reference level), as seen in Table 2. In the univariate model, average number of duplication events in AD−P subjects was 19.7, compared with them, average number of duplication events in AD+P subjects was marginally significantly lower (beta coefficient=−3.0, P=0.072); average number of duplications was still lower in AD+P than in AD−P (beta coefficient=−3.2, P=0.059) group in the multivariable model even after adjusting for age, sex and the first four principal components of population substructure.
CNV association analyses
Duplication CNVs
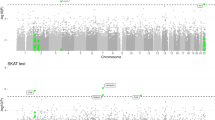
We conducted a genome-wide scan for CNVs associated with AD+P. The genome-wide P-values for duplication CNVs are shown in a Manhattan plot (Figure 1). We identified a duplication CNV on chromosome 19 that was genome-wide significantly protective against AD+P (odds ratio=0.42, P=7.20E−10 for the test based on ordered logistic regression). Forty-three out of 440 AD+P subjects carried this duplication (frequency=9.8%), while 172 out of 593 AD intermediate P and 33 out of 126 AD−P subjects had this duplication (frequency=29.0 and 24.3%, respectively). Although the duplication frequency is slightly higher in the intermediate group than in the AD−P subjects, the two groups are very similar and we interpret this departure from an ordered model as a function of the small sample size. It is worth noting that this result was highly significant even under the ordered model that we fit. This was a hemizygous duplication (copy number of 3). It was located on chromosome 19p13.3 and affected only one gene, APC2 (adenomatosis polyposis coli 2). Examples of this duplication in one CNV carrier and one subject without CNV based on LRR and BAF are shown in Figure 2. In addition, we detected four other duplications in which frequency was significantly lower (P<1.0E−05) in AD+P cases compared with non AD+P subjects. One was located on chromosome 16 and affected the ZFPM1 (zinc finger protein, FOG family member 1) gene; for those who carry the duplication, the odds of being AD+P versus non AD+P was 0.44 times lower than for those without this duplication (P=2.13E−07). The second duplication was located on chromosome 14 and affected the JAG2 (jagged 2) gene; the odds ratio of having AD+P in CNV carriers versus non-carriers was 0.43 (P=5.01E−07). The third duplication was on chromosome 9 and affected the SET (SET nuclear oncogene) gene; the AD+P odds ratio was 0.44 (P=1.95E−06) in CNV carriers. The fourth duplication was on chromosome 17 with AD+P odds ratio of 0.49 (P=4.25E−06) and affected no known genes. Detailed information on all five AD+P associated duplications is provided in Table 3.

Manhattan plot shows the genome-wide P-values for duplication CNVs associated with AD+P. This Manhattan plot demonstrates the locations across the chromosomes of the human genome (horizontal axis) where associations between duplication calls at markers (dots) and AD+P are shown using −log10 P-values (vertical axis). The higher the dots are, the stronger the genetic associations are. The strongest signal is seen on chromosome 19. AD, Alzheimer's disease; CNV, copy-number variant; P, psychosis.


Examples of duplication on chr 19 in one CNV carrier (a) and one non CNV subject (b), and examples of deletion on chr 9 in one CNV carrier (c) and one non CNV subject (d) based on LRR and BAF. Each data point in the plots is a single marker (SNP or CNV marker). The x axis shows marker’s base pair position on chromosome 19, based on Human NCBI Build 36 (hg18). For each subject, the y axis on the top panel is the intensity data termed LRR and the bottom panel is the genotype data termed BAF. (a) Chromosome 19 duplication carrier. In the LRR plot, the expected log2R ratio is zero for normal copy of autosomes; increases in log R ratio were observed in arrowed region; in the BAF plot, the values at 0, 1/2 and 1 represent the expected positions of disomic AA, AB and BB genotypes, respectively. For a hemizygous duplication with copy number of 3, it would have a wider BAF split (at 1/3 and 2/3). However, BAF values at 1/3 and 2/3 were not observed in this case because this region is composed of homozygous SNP markers and a lot of CNV markers. (b) Subject without duplication. In the LRR plot, the arrowed region has LRR around zero; in the BAF plot, the values were clustered around 0, 1/2 and 1 as expected for normal copy number. (c) Chromosome 9 deletion carrier. In the LRR plot, the expected log2R ratio is zero for normal copy of autosomes; decreases in log R ratio were observed in arrowed region; in the BAF plot, the values at 0, 1/2 and 1 represent the expected positions of disomic AA, AB and BB genotypes, respectively. For a hemizygous deletion with copy number of 1, the BAF value on 1/2 disappeared. (d) Subject without chr 9 deletion. In the LRR plot, the arrowed region has LRR around zero; in the BAF plot, the values were clustered around 0, 1/2 and 1 as expected for normal copy number. BAF, B allele frequency; CNV, copy-number variant; LRR, log R ratio; SNP, single-nucleotide polymorphism.
Deletion CNVs
A Manhattan plot showing genome-wide P-values for association of AD+P with deletion CNVs is presented in Figure 3. None of the deletions is genome-wide significantly associated with AD+P. However, we identified two deletions that showed a marginal association with AD+P (P<1.0E−3). One was located on chromosome 4, which was more frequent in AD+P subjects (26.1%) than in AD intermediate P (19.6%) and AD−P (12.5%) subjects. There were no known genes in this deletion region. Another deletion we identified was located on chromosome 9p22.2 and was present only in AD+P subjects (P=8.87E−04); none of AD intermediate P or AD−P subjects had this deletion. This was a hemizygous deletion (copy number=1) and it affected only one gene, CNTLN (centlein, centrosomal protein). Detailed information on these two deletions is given in Table 3. Examples of this deletion in one CNV carrier and one subject without CNV based on LRR and BAF are shown in Figure 2.

Manhattan plot shows the genome-wide P-values for deletion CNVs associated with AD+P. This Manhattan plot demonstrates the locations across the chromosomes of the human genome (horizontal axis) where associations between deletion calls at markers (dots) and AD+P are shown using −log10P-values (vertical axis). The higher the dots are, the stronger the genetic associations are. The strongest signals are seen on chromosome 4 and chromosome 9. AD, Alzheimer's disease; CNV, copy-number variant; P, psychosis.
Discussion
Here we report the first genome-wide CNV study of AD+P. We detected a genome-wide significant association between a duplication and AD+P. This hemizygous duplication (copy number of 3) covers the APC2 (adenomatous polyposis coli 2) gene on chr1913.3 and is protective against developing AD+P (odds ratio=0.42, P=7.2E−10). The frequency of this duplication was 10% in AD+P compared with 29% in AD subjects with intermediate P and 24% in AD−P. Previously, a large duplication in this region on chr 19 containing APC2 has been reported at 26% in healthy white males,36 which is similar to what we have observed in subjects with intermediate P and in AD−P.
APC2, also called APCL, closely resembles APC in its overall domain structure and function.37 It is primarily expressed in the central nervous system and spans a genomic region over 20 kb. APC2 is expressed specifically and abundantly in brain.38 APC2 protein could bind to beta-catenin and deplete the intracellular beta-catenin pool,38 suggesting that APC2 may participate in the regulation of neuronal functions in association with beta-catenin. The Wnt (wingless)/β-catenin signaling pathway has been involved in a multitude of neuronal processes, including regulation of synaptogenesis, synapse specificity, axon guidance, dendrite development and overall brain development.39 Wnt/β-catenin signaling pathway has been implicated in the pathogenesis of several neurodevelopmental disorders, including SCZ, bipolar disorder and autism spectrum disorders,40 while psychosis is a key feature of SCZ and present in many cases of bipolar disorder. It has been demonstrated that altered levels and/or phosphorylation states of β-catenin and GSK-3 in brain are related with SCZ and may represent susceptibility loci for SCZ.41, 42 Furthermore, it has been suggested that changes in β-catenin and GSK-3 may represent one of the mechanisms through which antipsychotics are able to exert behavioral changes.43 It is possible that altered expression level of APC2 affects the development of psychosis through Wnt (wingless)/β-catenin signaling pathway.
Besides the interaction with β-catenin, animal models suggest that APC2 could have a role in AD. Kuczera et al. (2010)44 analyzed the effect of APC2 deletion on AD using a mouse model. They found that although loss of APC2 did not affect the course of pathology in an AD, APC2 cKO mice displayed impaired spatial memory and severely impaired extinction of fear memories. They further proposed that the mechanisms by which amyloid-β-pathology and loss of APC2 affect memory function might share similar mechanism of action. It is unclear whether this mechanism is related to the critical role of APC2 in axonal projection and the cytoskeletal regulation at leading edges in response to extracellular signals.45, 46 It is interesting to note that APC2 is associated with susceptibility to SCZ.47 Accumulated evidence has shown that many common SNPs associated with SCZ are shared with bipolar disorder and autism, and recently some overlap between SCZ and psychosis in AD was also observed in a large-scale genome-wide association study.26 It is plausible that the genetic sharing could extend from SNPs to CNVs.
We also identified duplications affecting three other genes, including SET (chr 9), JAG2 (chr14) and ZFPM1 (chr16), which were tentatively associated with AD+P as they did not achieve genome-wide significance.
The protein encoded by SET, also named I2PP2A, is an endogenous inhibitor of protein phosphatase-2A (PP2A), which is a key protein phosphatase in dephosphorylating tau.48 SET is widely expressed in different tissues and localizes primarily in the nucleus.49 In AD brains, the compromised PP2A activity in cytoplasm was shown to be caused by increased levels of SET and its translocation from nucleus to the cytoplasm in neurons.50, 51, 52 Transfection in PC12 cells53 demonstrated that SET induces hyperphosphorylation of tau, suggesting that SET may be involved in AD through the regulation of tau hyperphosphorylation. There is substantial evidence54 that AD+P is associated with increased phosphorylated microtubule-associated protein tau in the dorsolateral prefrontal cortex. This does not go against our findings that frequency of this duplication in AD+P (6.1%) was marginally lower (odds ratio=0.44, P=1.95E−06) than in AD intermediate P (16%) and AD+P (17.6%). Though it is usually expected that full gene duplications increase the gene expression/function, partial duplications may lead to gene products with new functions or dysfunctional gene products that would compete with and negatively regulate the normal gene products.
JAG2, located on chr14 q32, was first identified as the human brain expressed sequence tags homologous to Delta and Serrate.55 Delta and Serrate are ligands of Notch receptor in Drosophila, which can activate Notch and related receptors. Luo et al.55 found that JAG2 was involved in human Notch1 pathway of signal transduction. The Notch pathway is often regarded as a developmental pathway, but Notch proteins and ligands are also expressed and active in the adult brain. Notch is a key regulator of adult neural stem cells, and involved in the regulation of migration, morphology, synaptic plasticity and survival of immature and ageing neurons. Accumulated evidence shows that aberrant Notch signaling contributes to the development of AD.56 Notch may be implicated in familial AD through interaction with presenilin and APP in adult brain. Findings that Notch1 expression was markedly elevated in AD brains in comparison with controls raise the possibility that Notch1 has a role in terminally differentiated neurons and AD.57,58,59
ZFPM1 (Zinc finger protein, FOG family member 1) is expressed in human hematopoietic tissues as well as in adult cerebellum, stomach, testis, lymph node, liver and pancreas.60 It has a role in erythroid differentiation. The relationship between ZFPM1 and psychiatric disorders is unknown.
We did not identify deletions that were genome-wide significantly associated with AD+P. The strongest associated deletions were located on chromosomes 4 and 9. The deletion on chromosome 4 did not cover any known gene, while deletion on 9p22.2 was nearby CNTLN (also named C9orf39). The latter was an uncommon deletion that was present in 7 out of 440 AD+P patients, but not in 136 AD−P or 593 AD intermediate P subjects. CNTLN encodes a protein, called centlein, which was first identified in rat brain.61 An aphidicolin-inducible common fragile site FRA9G has been mapped to 9p22.2, within C9orf39 gene.62 Fragile sites are regions of the genome that are prone to mutation and epigenetic changes; hence, hot spots for genomic instability. Common fragile sites are a component of normal chromosome structure and are composed of unstable DNA stretches that form gaps and breaks on metaphase chromosomes after partial inhibition of DNA synthesis. Increasing evidence links genomic and epigenomic instability, including multiple fragile sites regions to neuropsychiatric diseases, including SCZ and autism.63 It has been observed that fragile sites are more frequent in SCZ and co-localize with SCZ-linked genes. Overlapping deletions in the general population have been previously demonstrated at hg18 genomic position: 17 250 170–19 296 630 with frequency of 10 in 6533 subjects64 and at (hg18 genomic position: 17 226 376–17 367 629) with frequency 1 out of 1557 controls.65 More detailed information on these deletions can be found in the database of genomic variants66 (http://projects.tcag.ca/variation/).
In conclusion, using the Illumina HumanOmni1-Quad BeadChip, we have identified a genome-wide significant duplication in the APC2 gene on chromosome 19 associated with AD+P (P=7.2E−10). We also found suggestive associations (P<5.0E−6) of three other duplication loci in SET, JAG2 and ZFPM1. None of the observed deletions were genome-wide significant. Our study has identified novel susceptible genes for psychosis in AD and suggests the possibility of extending the genetic sharing among psychiatric disorders (for example, SCZ, autism and AD+P) from SNPs to CNVs. However, the relationship of CNVs and the functional activity of these genes need to be examined by experiments. The interpretation of this study is also limited by the lack of replication data; large-scale independent studies are warranted to replicate the findings reported in this study.
References
Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 2006; 63: 168–174.
Kamboh MI, Demirci FY, Wang X, Minster RL, Carrasquillo MM, Pankratz VS et al. Genome-wide association study of Alzheimer's disease. Transl Psychiatry 2012; 2: e117.
Grupe A, Abraham R, Li Y, Rowland C, Hollingworth P, Morgan A et al. Evidence for novel susceptibility genes for late-onset Alzheimer′s disease from a genome-wide association study of putative functional variants. Hum Mol Genet 2007; 16: 865–873.
Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, Lince DH et al. A high-density whole-genome association study reveals that APOE is the major susceptibility gene for sporadic late-onset Alzheimer′s disease. J Clin Psychiatry 2007; 68: 613–618.
Reiman EM, Webster JA, Myers AJ, Hardy J, Dunckley T, Zismann VL et al. GAB2 alleles modify Alzheimer′s risk in APOE epsilon4 carriers. Neuron 2007; 54: 713–720.
Li H, Wetten S, Li L, St Jean PL, Upmanyu R, Surh L et al. Candidate single-nucleotide polymorphisms from a genomewide association study of Alzheimer disease. Arch Neurol 2008; 65: 45–53.
Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF et al. Genome-wide association analysis reveals putative Alzheimer′s disease susceptibility loci in addition to APOE. Am J Hum Genet 2008; 83: 623–632.
Beecham GW, Martin ER, Li YJ, Slifer MA, Gilbert JR, Haines JL et al. Genome-wide association study implicates a chromosome 12 risk locus for late-onset Alzheimer disease. Am J Hum Genet 2009; 84: 35–43.
Abraham R, Moskvina V, Sims R, Hollingworth P, Morgan A, Georgieva L et al. A genome-wide association study for late-onset Alzheimer′s disease using DNA pooling. BMC Med Genomics 2008; 1: 44.
Carrasquillo MM, Zou F, Pankratz VS, Wilcox SL, Ma L, Walker LP et al. Genetic variation in PCDH11X is associated with susceptibility to late-onset Alzheimer's disease. Nat Genet 2009; 41: 192–198.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer′s disease. Nat Genet 2009; 41: 1094–1099.
Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer′s disease. Nat Genet 2009; 41: 1088–1093.
Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010; 303: 1832–1840.
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer′s disease. Nat Genet 2011; 43: 429–435.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer′s disease. Nat Genet 2011; 43: 436–441.
Guffanti G, Torri F, Rasmussen J, Clark AP, Lakatos A, Turner JA et al. Increased CNV-region deletions in mild cognitive impairment (MCI) and Alzheimer's disease (AD) subjects in the ADNI sample. Genomics 2013; 102: 112–122.
Szigeti K, Kellermayer B, Lentini JM, Trummer B, Lal D, Doody RS et al. Ordered subset analysis of copy number variation association with age at onset of Alzheimer's disease. J Alzheimers Dis 2014; 41: 1063–1071.
Swaminathan S, Shen L, Kim S, Inlow M, West JD, Faber KM et al. Analysis of copy number variation in Alzheimer's disease: the NIALOAD/ NCRAD Family Study. Curr Alzheimer Res 2012; 9: 801–814.
Szigeti K, Lal D, Li Y, Doody RS, Wilhelmsen K, Yan L et al. Genome-wide scan for copy number variation association with age at onset of Alzheimer's disease. J Alzheimers Dis 2013; 33: 517–523.
Ghani M, Pinto D, Lee JH, Grinberg Y, Sato C, Moreno D et al. Genome-wide survey of large rare copy number variants in Alzheimer's disease among Caribbean hispanics. G3 (Bethesda) 2012; 2: 71–78.
Rovelet-Lecrux A, Legallic S, Wallon D, Flaman JM, Martinaud O, Bombois S et al. A genome-wide study reveals rare CNVs exclusive to extreme phenotypes of Alzheimer disease. Eur J Hum Genet 2012; 20: 613–617.
Chapman J, Rees E, Harold D, Ivanov D, Gerrish A, Sims R et al. A genome-wide study shows a limited contribution of rare copy number variants to Alzheimer's disease risk. Hum Mol Genet 2013; 22: 816–824.
Shaw CA, Li Y, Wiszniewska J, Chasse S, Zaidi SN, Jin W et al. Olfactory copy number association with age at onset of Alzheimer disease. Neurology 2011; 76: 1302–1309.
Heinzen EL, Need AC, Hayden KM, Chiba-Falek O, Roses AD, Strittmatter WJ . Genome-wide scan of copy number variation in late-onset Alzheimer's disease. J Alzheimers Dis 2010; 19: 69–77.
Bacanu SA, Devlin B, Chowdari KV, DeKosky ST, Nimgaonkar VL, Sweet RA . Heritability of psychosis in Alzheimer disease. Am J Geriatr Psychiatry 2005; 13: 624–627.
Hollingworth P, Sweet R, Sims R, Harold D, Russo G, Abraham R et al. Genome-wide association study of Alzheimer's disease with psychotic symptoms. Mol Psychiatry. 2012; 17: 1316–1327.
Lopez OL, Becker JT, Klunk W, Saxton J, Hamilton RL, Kaufer D et al. Research evaluation and diagnosis of probable Alzheimer's disease over the last two decades: I. Neurology 2000; 55: 1854–1862.
Lopez OL, Becker JT, Klunk W, Saxton J, Hamilton RL, Kaufer D et al. Research evaluation and diagnosis of possible Alzheimer's disease over the last two decades: II. Neurology 2000; 55: 1863–1869.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM . Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984; 34: 939–944.
Tariot PN, Mack JL, Patterson MB, Edland SD, Weiner MF, Fillenbaum G et al. The Behavior Rating Scale for Dementia of the Consortium to Establish a Registry for Alzheimer’s Disease. Am J Psychiatry 1995; 152: 1349–1357.
DeMichele-Sweet MA, Klei L, Devlin B, Ferrell RE, Weamer EA, Emanuel JE . No association of psychosis in Alzheimer disease with neurodegenerative pathway genes. Neurobiol Aging 2011; 32: 555.e9–555.e11.
Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF . PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007; 17: 1665–1674.
Willis JA, Mukherjee S, Orlow I, Viale A, Offit K, Kurtz RC et al. Genome-wide analysis of the role of copy-number variation in pancreatic cancer risk. Front Genet 2014; 5: 29.
Elia J, Glessner JT, Wang K, Takahashi N, Shtir CJ, Hadley D et al. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat Genet 2011; 44: 78–84.
R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2012 ISBN 3-900051-07-0, URL http://www.R-project.org.
de Smith AJ, Tsalenko A, Sampas N, Scheffer A, Yamada NA, Tsang P et al. Array CGH analysis of copy number variation identifies 1284 new genes variant in healthy white males: implications for association studies of complex diseases. Hum Mol Genet 2007; 16: 2783–2794.
van Es JH, Kirkpatrick C, van de Wetering M, Molenaar M, Miles A, Kuipers J et al. Identification of APC2, a homologue of the adenomatous polyposis coli tumour suppressor. Curr Biol 1999; 9: 105–108.
Nakagawa H, Murata Y, Koyama K, Fujiyama A, Miyoshi Y, Monden M et al. Identification of a brain-specific APC homologue, APCL, and its interaction with beta-catenin. Cancer Res 1998; 58: 5176–5181.
Freyberg Z, Ferrando SJ, Javitch JA . Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiatry 2010; 167: 388–396.
Okerlund ND, Cheyette BN . Synaptic Wnt signaling-a contributor to major psychiatric disorders? J Neurodev Disord 2011; 3: 162–174.
Mao Y, Ge X, Frank CL, Madison JM, Koehler AN, Doud MK et al. Disrupted in schizophrenia 1 regulates neuronal progenitor proliferation via modulation of GSK3beta/beta-catenin signaling. Cell 2009; 136: 1017–1031.
Singh KK . An emerging role for Wnt and GSK3 signaling pathways in schizophrenia. Clin Genet 2013; 83: 511–517.
Alimohamad H, Rajakumar N, Seah YH, Rushlow W . Antipsychotics alter the protein expression levels of beta-catenin and GSK-3 in the rat medial prefrontal cortex and striatum. Biol Psychiatry 2005; 57: 533–542.
Kuczera T, Stilling RM, Hsia HE, Bahari-Javan S, Irniger S, Nasmyth K et al. The anaphase promoting complex is required for memory function in mice. Learn Mem 2010; 18: 49–57.
Shintani T, Takeuchi Y, Fujikawa A, Noda M . Directional neuronal migration is impaired in mice lacking adenomatous polyposis coli 2. J Neurosci 2012; 32: 6468–6484.
Shintani T, Ihara M, Tani S, Sakuraba J, Sakuta H, Noda M . APC2 plays an essential role in axonal projections through the regulation of microtubule stability. J Neurosci 2009; 29: 11628–11640.
Cui DH, Jiang KD, Jiang SD, Xu YF, Yao H . The tumor suppressor adenomatous polyposis coli gene is associated with susceptibility to schizophrenia. Mol Psychiatry 2005; 10: 669–677.
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX . Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 2005; 22: 1942–1950.
Tanimukai H, Grundke-Iqbal I, Iqbal K . Inhibitors of protein phosphatase-2A: topography and subcellular localization. Brain Res Mol Brain Res 2004; 126: 146–156.
Tsujio I, Zaidi T, Xu J, Kotula L, Grundke-Iqbal I, Iqbal K . Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett 2005; 579: 363–372.
Tanimukai H, Grundke-Iqbal I, Iqbal K . Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer's disease. Am J Pathol 2005; 166: 1761–1771.
Yu G, Yan T, Feng Y, Liu X, Xia Y, Luo H et al. Ser9 phosphorylation causes cytoplasmic detention of I2PP2A/SET in Alzheimer disease. Neurobiol Aging 2013; 34: 1748–1758.
Chohan MO, Khatoon S, Iqbal IG, Iqbal K . Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett 2006; 580: 3973–3979.
Murray PS, Kumar S, Demichele-Sweet MA, Sweet RA . Psychosis in Alzheimer's disease. Biol Psychiatry 2014; 75: 542–552.
Luo B, Aster JC, Hasserjian RP, Kuo F, Sklar J . Isolation and functional analysis of a cDNA for human Jagged2, a gene encoding a ligand for the Notch1 receptor. Mol Cell Biol 1997; 17: 6057–6067.
Kyriazis GA, Belal C, Madan M, Taylor DG, Wang J, Wei Z et al. Stress-induced switch in Numb isoforms enhances Notch-dependent expression of subtype-specific transient receptor potential channel. J Biol Chem 2010; 285: 6811–6825.
Song W, Nadeau P, Yuan M, Yang X, Shen J, Yankner BA . Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci USA 1999; 96: 6959–6963.
Berezovska O, Xia MQ, Hyman BT . Notch is expressed in adult brain, is coexpressed with presenilin-1, and is altered in Alzheimer disease. J Neuropathol Exp Neurol 1998; 57: 738–745.
Ables JL, Breunig JJ, Eisch AJ, Rakic P . Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci 2011; 12: 269–283.
Freson K, Thys C, Wittewrongel C, Vermylen J, Hoylaerts MF, Van Geet C . Molecular cloning and characterization of the GATA1 cofactor human FOG1 and assessment of its binding to GATA1 proteins carrying D218 substitutions. Hum Genet 2003; 112: 42–49.
Makino K, Umeda K, Uezu A, Hiragami Y, Sakamoto T, Ihn H et al. Identification and characterization of the novel centrosomal protein centlein. Biochem Biophys Res Commun 2008; 366: 958–962.
Sawińska M, Schmitt JG, Sagulenko E, Westermann F, Schwab M, Savelyeva L . Novel aphidicolin-inducible common fragile site FRA9G maps to 9p22.2, within the C9orf39 gene. Genes Chromosomes Cancer 2007; 46: 991–999.
Smith CL, Bolton A, Nguyen G . Genomic and epigenomic instability, fragile sites, schizophrenia and autism. Curr Genomics 2010; 11: 447–469.
Xu H, Poh WT, Sim X, Ong RT, Suo C, Tay WT et al. SgD-CNV, a database for common and rare copy number variants in three Asian populations. Hum Mutat 2011; 32: 1341–1349.
Itsara A, Cooper GM, Baker C, Girirajan S, Li J, Absher D et al. Population analysis of large copy number variants and hotspots of human genetic disease. Am J Hum Genet 2009; 84: 148–161.
MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW . The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014; 42 (Database issue): D986–D992.
Acknowledgements
This study was supported by the National Institute on Aging grants AG030653, AG041718 (MIK), AG005133 (OLL) and AG027224 (RAS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Department of Veterans Affairs or the United States Government. This work was conducted in partial fulfillment of requirements for the first author’s doctoral degree, supported by NIMH Grant T32 MH015169 (GAR, Program Director).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Zheng, X., Demirci, F., Barmada, M. et al. Genome-wide copy-number variation study of psychosis in Alzheimer’s disease. Transl Psychiatry 5, e574 (2015). https://doi.org/10.1038/tp.2015.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/tp.2015.64
This article is cited by
-
DNA methylation signatures of Alzheimer’s disease neuropathology in the cortex are primarily driven by variation in non-neuronal cell-types
Nature Communications (2022)
-
Psychosis in Alzheimer’s Disease
Current Neurology and Neuroscience Reports (2020)
-
GBA and APOE ε4 associate with sporadic dementia with Lewy bodies in European genome wide association study
Scientific Reports (2019)
-
Revealing Alzheimer’s disease genes spectrum in the whole-genome by machine learning
BMC Neurology (2018)
-
Network biology concepts in complex disease comorbidities
Nature Reviews Genetics (2016)
