
Abstract
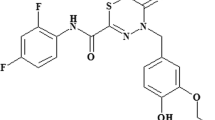
Therapeutic strategies that target bacterial virulence have received considerable attention. The type III secretion system (T3SS) is important for bacterial virulence and represents an attractive therapeutic target. A novel compound with a predicted T3SS inhibitory activity named CL-55 (N-(2,4-difluorophenyl)-4-(3-ethoxy-4-hydroxybenzyl)-5-oxo-5,6-dihydro-4H-[1,3,4]-thiadiazine-2-carboxamide) was previously characterized by low toxicity, high levels of solubility, stability and specific efficiency toward Chlamydia trachomatis in vitro and in vivo. In this study, we describe the action of CL-55 on Salmonella enterica serovar Typhimurium. We found that CL-55 does not affect Salmonella growth in vitro but suppresses Salmonella infection in vivo. The i.p. injection of CL-55 at a dose of 10 mg kg−1 for 4 days significantly (500-fold) decreased the numbers of Salmonella in the spleen and peritoneal lavages and increased the survival rates in susceptible (BALB/c, I/St) and resistant (A/Sn) mice. Twelve days of therapy led to complete eradication of Salmonella in mice. Moreover, no pathogen was found 4–6 weeks post treatment. CL-55 was not carcinogenic or mutagenic, did not increase the level of chromosomal aberrations in bone marrow cells and had low toxicity in mice, rats and rabbits. Pharmacokinetic studies have shown that CL-55 rapidly disappears from systemic blood circulation and is distributed in the organs. Our data demonstrates that CL-55 affects S. enterica serovar Typhimurium in vivo and could be used as a substance in the design of antibacterial inhibitors for pharmaceutical intervention of bacterial virulence for infection.
Similar content being viewed by others
Introduction
Salmonella enterica is an important pathogen of humans and animals. This pathogen can cause diseases ranging from mild gastroenteritis to severe systemic infection and is a serious problem for public health. Recently we observed outbursts of infection that lead to generalized salmonellosis, which in turn results in chronic complications such as gastroenteritis, colitis, cholecystitis, mesenchymal hepatitis, nephritis, arthritis, periostitis and perichondritis.1 These diseases are difficult to treat with antibiotics. In addition, antibiotic-resistant strains are constantly emerging worldwide, posing a serious problem especially for newborns and children. Thus, the demand for novel antimicrobial strategies to replenish the dwindling antibiotic arsenal has increased.
Therapeutic strategies that target bacterial virulence rather than growth have received considerable attention as these approaches are aimed to selectively disarm pathogens while preserving the integrity of the host microbiome. Compounds that inhibit the secretion of virulence factors and suppress the pathogenicity of bacteria normally do not affect viability. Eradication of the pathogen that lost its virulence occurs as the result of natural resistance and immune response from the host. As virulence inhibitors do not cause the death and do not suppress the growth of the pathogen, this therapy should not lead to the selection of resistant bacteria. Amongst the many bacterial virulence mechanisms that have been discovered, specialized protein secretion systems appear to be the prime targets for small-molecule inhibition of infection. At least seven secretion systems are known at present. The highly conserved type III secretion system (T3SS) is central to the virulence of many human Gram-negative pathogens such as Salmonella, Shigella, Pseudomonas, enteropathogenic Escherichia coli, enterohemorrhagic E. coli, Vibrio, Yersinia and Chlamydia.2, 3, 4, 5, 6 The pathogenicity of S. enterica mainly depends on two T3SSs, which are encoded by Salmonella pathogenicity island 1 (SPI-1) and SPI-2. SPI-1 mediates the invasion of the intestinal epithelium and induction of proinflammatory responses through injecting effector proteins into host cells, while SPI-2 regulates the replication of bacteria in host phagocytic cells.7 T3SSs have therefore emerged as attractive targets for small-molecule anti-virulence therapeutics and have motivated several high-throughput screens in the search for specific chemical inhibitors that inhibit the secretion and translocation of bacterial effector proteins. Several series of T3SS inhibitors have been identified by whole-cell-based screening methods, such as reporter gene screening system, enzyme-linked immunosorbent assay, contact hemolysis screening system and SDS-polyacrylamide gel electrophoresis.8
The salicylidene acylhydrazides were the first class of small molecules identified from a T3SS effector screen. The compounds of this class were shown to have T3SS inhibitory activity in several Gram-negative pathogens including E. coli,9, 10 Chlamydia,11, 12 Salmonella13, 14, 15 and Shigella16 that led to attenuation of infection.
The therapeutic effect of T3SS inhibitors in vivo has been described in a few studies. A decrease in inflammation and bacterial load of S. enterica serovar Typhimurium,17 Yersinia pseudotuberculosis18 and Chlamydia trachomatis19 but only as prophylactic treatment before infection was shown in previous studies. Eradication of Citrobactor rodentium in the model of colitis under the action of compound aurodox, therapy of which was started immediately after infection, was also reported;20 however, inhibiting the action of aurodox on T3SS was demonstrated for enteropathogenic E. coli, but not for C. rodentium, and the mechanism of the action of this compound on the course of C. rodentium infection is not clear.
Previously, our group tested 500 compounds of thiohydrazides of oxamic acids with predicted T3SS inhibitory activity for toxicity to eukaryotic cells and antibacterial inhibitory activity. The selected T3SS inhibitors were chemically optimized, which allowed us to obtain the original lead compound named CL-55, characterized by low toxicity, high levels of solubility, stability and specific efficiency on Chlamydia in vitro and in vivo.21, 22
In this study we describe the antibacterial action of CL-55 (N-(2,4-difluorophenyl)-4-(3-ethoxy-4-hydroxybenzyl)-5-oxo-5,6-dihydro-4H-[1,3,4]-thiadiazine-2-carboxamide) on S. enterica serovar Typhimurium. We found that CL-55 does not affect Salmonella growth in vitro but suppresses Salmonella infection in vivo. We found that CL-55 significantly decreased mortality and bacteria loads in susceptible and resistant mice infected with S. enterica serovar Typhimurium. In addition, we demonstrated that CL-55 was not carcinogenic or mutagenic, did not increase the level of chromosomal aberrations in bone marrow cells and displayed low toxicity in mice, rats and rabbits. Preclinical pharmacokinetics studies showed fast access of CL-55 into the bloodstream via i.p. injection and its distribution into body organs. Our data demonstrate that the selected compound CL-55 affects S. enterica serovar Typhimurium infection in vivo and hence could be used as a substance in the design of bacterial T3SS specific inhibitors for pharmaceutical intervention of bacterial virulence.
Materials and methods
Bacterial strains and cultivation conditions
This study was based on the S. enterica serovar Typhimurium strain IV147 obtained from H. Tschape from the Robert Koch Institute branch at Wernigerode, Germany. Bacteria were grown in Luria-Bertani broth (1% tryptone, 0.5% yeast extract, 1% NaCl, pH 7.4) or on Salmonella Shigella (SS) agar plates.
CL-55 compound
The virulence inhibitor CL-55 (Figure 1) was synthesized as described earlier.20 In our study several solutions of inhibitor were used. A 100 mm stock solution in dimethyl sulfoxide was used for the in vitro experiments. A solution for in vivo studies was prepared in the following way: 2 mg of CL-55 were dissolved in 200 μl of 0.1 m NaOH, then 400 μl of 0.05 N AcONH4 were added. The resulting solution was acidified with AcOH up to pH 6.8–7.2 and then diluted with distilled water up to 1 ml. The suspensions for the toxicity studies were prepared in the following way: 100 mg of CL-55 were thoroughly mixed with 50 μl of Tween-80, then diluted with 1% starch solution up to 1 ml. The solution for pharmacokinetics was prepared by dissolving of 40 mg CL-55 in 1 ml of PEG 400 (polyethylene glycol-400). All of the solutions were prepared ex tempera.

The structure of compound СL-55 (N-(2,4-difluorophenyl)-4-(3-ethoxy-4-hydroxybenzyl)-5-oxo-5,6-dihydro-4H-[1,3,4]-thiadiazine-2-carboxamide; Patent RU 2495036 C1).
Animals
Male mice of 14–16 g body weight of I/StSnEgYCit (I/St) and A/JSnYCit (A/Sn) and BALB/cJCit (BALB/c) inbred strains were used for the evaluation of CL-55 efficiency in a Salmonella infection model in vivo. Male and female F1CBAхC57Bl/6 mice were used in the mutagenicity studies. BALB/c mice, Wistar rats and Shinshilla rabbits were used in the toxicity studies. BALB/c mice were also used in the pharmacokinetics study.
The animals were maintained under conventional conditions at the Animal Facilities of the Gamaleya Research Institute for epidemiology and microbiology, Moscow, Russia, in accordance with national guidelines. Water and food were provided ad libitum.
Acute infection model in vivo
Acute Salmonella infection was induced as described previously.23 Briefly, S. enterica serovar Typhimurium (var. IV147) was grown in broth overnight, centrifuged, resuspended in sterile saline and 0.5 ml (2.5 × 102 CFU per mouse) were injected i.p. Mortality was monitored daily. To estimate the Salmonella loads, serial dilutions of peritoneal cavity lavages and whole-spleen homogenates were plated onto SS agar. The colonies were counted following a 12 h incubation at 37 °C.
Mutagenicity and carcinogenicity of CL-55
Ames test
It was performed according to a commercial protocol of the ‘AMES MPF PENTA I’ kit (Xenometrix AG, Allschwil, Switzerland). S. enterica serovar Typhimurium strains ТА98, ТА100, ТА1535, ТА1537 and E. coli strains pKM101 и uvrA were used in this study. The compound CL-55 was evaluated at concentrations of 0.00032, 0.0016, 0.008, 0.04, 0.2 and 1 mg ml−1.
Comet assay
It was performed in the alkaline version as described before.24 CL-55 was administered as a single dose of 20 or 200 mg kg−1 in 1% Tween-80 solution i.m. The samples of spleen, liver and kidney cells were prepared 3 and 18 h later. A 1% solution of Tween-80 was used as the negative control. The i.p. injection of 40 mg kg−1 methyl methane sulfonate was used as a positive control.
The formation of chromosomal aberrations in the bone marrow cells
The formation of chromosomal aberrations in the bone marrow cells was evaluated 24 h as described before25 after i.m. administration of CL-55 at doses of 20 and 200 mg kg−1 and after repeated daily i.m. injections of 20 mg kg−1 of CL-55 for 5 days. The samples were collected 6 h after the last injection. A 1% solution of Tween-80 was used as a negative control. The i.p. injection of 20 mg kg−1 cyclophosphamide (Biohimic, Saransk, Russia) was used as a positive control.
LD50 estimation
Acute toxicity was studied after i.p. injections of CL-55 to animals in a single dose from 500 to 5000 mg kg−1 in starch with Tween-80. Macroscopic examination and morphometric evaluation of the organs were evaluated at day 14 after the treatment.
Subchronic toxicity
Subchronic toxicity was studied after per os injection of CL-55 to animals in a single daily dose of 50 or 500 mg kg−1 in starch with Tween-80 for 14 days. The physiological parameters such as blood counts and biochemistry, urine tests and organ histology were evaluated in this study at 24 h and 28 days after the end of the treatment.
Pharmacokinetics
The mice were injected i.p. with 100 mg kg−1 of CL-55 in PEG 400. Blood samples were collected in heparin-treated tubes at 5, 10, 15, 30, 60 and 120 min post injection. Blood plasma was separated by centrifugation, diluted with acetonitrile and cleared again by centrifugation.
Liquid UV chromatography of compound concentration in blood and organs
Concentrations of the compound were analyzed using a liquid chromatography UV spectrometry system Gilson UV–VIS Detector 155 with two pumps (Gilson 305, Gilson 306), a system controller Gilson 506 C (Gilson, Middleton, WI, USA) and Hitachi 5210 autosampler (Hitachi High-Tech Corp., Tokyo, Japan). The analytical separation column (Reprosil Pur-Basic 250 × 4.6 mm 5 μm (Dr Maisch, Germany) was coupled with a pre-column (Security Guard Cartridge C18 4 × 3 mm, 5 μm). An isocratic elution of the mobile phase (MeCN/TFA/water 60/0.5/39.5) was run with a total run time of 10 min, a flow rate of 1.5 ml min−1, and a detection wave of 281 nm.
The samples were collected by the complete loop filling method (50 μl). The accuracy ranged from 90 to 110%, and the precision ranged from ±10% for the standard concentration (250–15 000 ng ml−1) except for the lower limit of quantification (100 ng ml−1), where the accuracy was 80–120% and precision ±20%. The limits of detection (LOD) was 25 ng ml−1. The concentration of CL-55 in the samples was determined by comparing the peak areas of the compounds with a standard calibration curve.
Pharmacokinetic modeling
Non-compartment analysis of plasma/whole-blood concentration was performed using the Microsoft Excel-based program PK Solver 2.0. The area under the zero moment (AUC0–t) and first moment curves (AUMC0–t) from 0 to the last time t were calculated using the linear trapezoidal method. The terminal elimination slope (lz) was estimated using regression with the largest R2 on the points before peak plasma concentration (Cmax). The elimination half-life (t1/2), volume of distribution to fractional bioavailability (Vd/F), clearance to fractional bioavailability (Cl/F) and mean residence time parameters were also determined. Peak plasma drug concentrations (Cmax) and the times to reach the peak concentration (Tmax) values were derived from the plasma concentration versus the time profiles.
Statistics
The results obtained from the mortality rates studies are represented as Kaplan–Meier survival curves, and the differences in survival were calculated by the log-rank test. Significant differences of the other data were determined using the Mann–Whitney non-parametric two-tailed test using GraphPad Prism Version 6.
Results
CL-55 efficiently inhibits acute S. enterica serovar Typhimurium infection in vivo
Initially, we examined whether CL-55 can affect the growth of S. enterica serovar Typhimurium IV147 in Luria-Bertani broth. The overnight culture IV 147 strain was diluted with fresh broth 10 times and the bacteria were treated with a range of CL-55 concentrations (50, 150 and 200 μм) or diluent and incubated at 37 °С for 24 h. The number of living bacteria was counted by plating from serial dilutions of cultures on SS agar.
The number of Salmonella was 2 × 109 CFU per ml for the CL-55-treated cultures and control. We did not find a significant difference in the rate of bacteria growth between the CL-55-treated or control cultures and demonstrated that CL-55 did not affect the growth of Salmonella in vitro at the given concentrations.
To analyze the action of CL-55 on the course of Salmonella infection in vivo we used mouse strains with different levels of susceptibility to Salmonella—highly susceptible (Nramp1s) BALB/c mice, susceptible I/St mice and resistant A/Sn mice (both strains are Nramp1r). The mice with different levels of susceptibility to infection allowed us to model the variability of response to Salmonella infection in humans.
The mice were infected with a dose of 2.5 × 102 CFU per mouse of virulent Salmonella via the i.p. route. CL-55 was administered at three different i.p. doses (1.0, 10 and 40 mg kg−1) for 4 days starting from day 1 post infection. The mortality and time course of bacteria loads in the different organs were evaluated in these experiments.
First, we evaluated the effect of CL-55 on i.p. Salmonella infection in BALB/c mice. Injection of 1.0 mg kg−1 of CL-55 for 4 days starting from day 1 post infection did not affect the survival rates in BALB/c mice infected with Salmonella, whereas the mice that received 10 (Figure 2a) and 40 mg kg−1 (not shown) of CL-55 had an extended survival rate compared with the mice that did not receive the treatment (P<0.001; Figure 2a). The mean survival rate in susceptible BALB/c mice that received 10 mg kg−1 was 25±1.2 days compared with 12±1.5 days in the control group (Figure 2a). These results suggest that CL-55 injected to susceptible BALB/c mice at doses of 10 and 40 mg kg−1 during 4 days post infection displayed a pronounced antibacterial activity in the model of acute Salmonella infection in vivo.

CL-55 (N-(2,4-difluorophenyl)-4-(3-ethoxy-4-hydroxybenzyl)-5-oxo-5,6-dihydro-4H-[1,3,4]-thiadiazine-2-carboxamide) increases the survival rates of BALB/cJCit (a), A/JSnYCit (b) and I/StSnEgYCit (c) mice with acute Salmonella infection. Mice were infected with Salmonella via the i.p. route and treated with 10 mg kg−1 of CL-55 for 4 days from the day of infection (dashed line). Infected, untreated mice were used as controls. Representative data of one of three experiments is presented.
Next, we analyzed the activity of CL-55 on acute Salmonella infection in A/Sn and I/St mice. A/Sn mice are more resistant to Salmonella, respiratory Chlamydia and tuberculosis infection than I/St mice.23 The genetic mechanism of susceptibility of I/St mice differs from the susceptibility of BALB/c mice as A/Sn and I/St mice do not have a mutation in the Nramp1 gene.23
The medium time of survival of resistance to Salmonella for the A/Sn mice treated with 10 mg kg−1 of CL-55 was 28±1.5 days, whereas the control (non-treated) mice survived for 15±1.2 days (P<0.001). The medium time of survival of susceptibility to Salmonella for the I/St mice treated with CL-55 was 15±0.8 days, whereas the control (non-treated) mice survived for 8±0.9 days (P<0.001; Figures 2b and c). Therefore, in this study we showed that CL-55 had antibacterial activity in the model of acute salmonellosis in three strains of inbred mice differing in susceptibility of Salmonella.
In addition to mortality studies, we have analyzed the course of bacterial multiplication in the peritoneal cavity and spleen of mice treated with 10 mg kg−1 of CL-55 for 4 days starting from day 1 post infection. To this end, treated or control mice were sacrificed at days 1 to 3 after treatment, the spleen and peritoneal cavity samples were cultured in SS agar and the numbers of live Salmonella were assessed by counting the CFU. The results are presented in Figure 3.

CL-55 (N-(2,4-difluorophenyl)-4-(3-ethoxy-4-hydroxybenzyl)-5-oxo-5,6-dihydro-4H-[1,3,4]-thiadiazine-2-carboxamide) decreases Salmonella numbers in the spleens and peritoneal cavities in BALB/cJCit (BALB/c) mice. BALB/c mice were infected i.p. with Salmonella and treated with 10 mg kg−1 of CL-55 for 4 days starting from day 1 post infection. Round markers—Salmonella loads in the spleens of untreated mice; triangle markers—Salmonella loads in the peritoneal cavities of untreated mice; large square markers—Salmonella loads in the spleens of CL-55 treated mice; small square markers—Salmonella loads in the peritoneal cavities of CL-55-treated mice. The mean±s.d. are presented. *P-value <0.001.
As shown in Figure 3, treatment with CL-55 resulted in a significant (~500-fold, P<0.001) decrease in the Salmonella numbers in the peritoneal lavages and spleens compared with the Salmonella numbers in the organs of non-treated animals.
As CL-55 did not affect the growth of Salmonella in broth in culture, the decrease of bacterial loads shown in Figure 3 is likely due to host natural resistance mechanisms activated as a result of the loss of bacterial virulence.
As we showed that 4 days of therapy decreased the mortality of Salmonella-infected mice, we next evaluated the efficiency after 12 days of therapy with 10 mg kg−1 of CL-55 in BALB/c mice. We chose this duration of therapy according to the survival time of the infected mice that did not receive treatment.
We found that treatment of BALB/c mice with 10 mg kg−1 of CL-55 for 12 days resulted in 100% survival, whereas all of the untreated mice were dead at day 12 post infection. The results are shown in Figure 4. Notably, the mice treated with CL-55 for 12 days survived for an additional 2 months of observation. The spleens obtained from these mice at 4–6 weeks post treatment were evaluated for CFU counts. We found that the spleens were Salmonella negative, confirming the absence of pathogen reactivation after the treatment with our inhibitor (data not shown).

The treatment of BALB/cJCit mice with 10 mg kg−1 of CL-55 (N-(2,4-difluorophenyl)-4-(3-ethoxy-4-hydroxybenzyl)-5-oxo-5,6-dihydro-4H-[1,3,4]-thiadiazine-2-carboxamide) for 12 days resulted in 100% survival, whereas all of the untreated mice were dead at day 12 post infection. Mice were infected with Salmonella via the i.p. route and treated with 10 mg kg−1 of CL-55 for 4 days from the day of infection (dashed line). Infected, untreated mice were used as controls. Representative data of one of the three experiments is presented.
The data obtained in our experiments suggest that CL-55 possesses significant antibacterial activity in the model of acute Salmonella infection. Twelve days of treatment with 10 mg kg−1 of CL-55 resulted in 100% survival of the animals, eradication of infection from the organs and lack of infection reactivation for an additional 2 months post infection.
Acute and subchronic toxicity of CL-55 compound
We did not reach the LD50 due to the low toxicity of CL-55. The maximum dose injected into mice and rats was 5000 mg kg−1. A single dose of 5000 mg kg−1 did not lead to mortality or toxic changes in the organs of animals (Supplementary Tables 2–5).
Multiple per os administration of CL-55 at a dose of 50 mg kg−1 for 14 days to rats and rabbits (Supplementary Tables 6 and 7) were not toxic for the animals.
The administration of CL-55 at a dose of 500 mg kg−1 to rats and rabbits revealed some negative changes in the structure of the thymus (data not shown) in rats and showed a decrease in alanine transferase in the serum as well as a decrease in sodium, creatinine and urea in both of the animal species (Supplementary Table 6). All of the pathological findings were reversible. The results of our study suggest that CL-55 has negligible toxicity.
Evaluation of potential mutagenicity and carcinogenicity of CL-55
As CL-55 showed good anti-bactericidal potential in vitro and in vivo, we next analyzed the mutagenicity and carcinogenic potential of the compound. To this end, we employed an Ames test. CL-55 had no mutagenic effect on TA98, TA100, TA1535 and TA1537 strains of S. enterica serovar Typhimurium and pKM101 and uvrA strains of E. coli at all concentrations of the compound in the Ames test with or without metabolic activation. The number of reverse mutants in the controls was within the level of spontaneous fluctuations for these strains (Supplementary Table 11).
CL-55 (20 or 200 mg kg−1) injected i.m. into mice did not induce an increase in DNA repairs in bone marrow, liver, kidney and spleen cells when tested in a Comet assay 18 h after injection compared with the negative control, while injection of methyl methane sulfonate induced a statistically significant increase in DNA repairs in all of the organs under study. (Supplementary Table 12).
The study of chromosomal aberrations in bone marrow cells showed that a single injection of 20 or 200 mg kg−1 of CL-55 resulted in a chromosomal aberrations level of 1.0±0.4% and 1.6±0.6% of the analyzed cells, respectively. The level of chromosomal aberrations in male mice after five injections of 20 mg ml−1 of CL-55 was 1.2±0.5%. Female mice injected with CL-55 at a dose of 20 mg kg−1 had 1.6±0.6% chromosomal aberrations. The spontaneous level of chromosomal aberrations in bone marrow cells was 0.8±0.4% (Supplementary Table 13). Thus, neither a single nor repeated dose of 20 mg kg−1 of CL-55-induced chromosomal aberrations in the bone marrow of male and female mice. These results demonstrate that CL-55 does not have mutagenic or potentially carcinogenic activity.
Preclinical pharmacokinetics in mice
CL-55 injected i.p. at a dose of 100 mg kg−1 in a volume of 50 μl was chosen for evaluation of the pharmacokinetic parameters in mice. The blood plasma analysis showed that CL-55 reached a Cmax (10.98 μg ml−1) in the plasma 10 min after injection (Table 1) and disappeared quickly from the bloodstream within 120 min post injection (Figure 5). The high value of the volume of distribution (Vz/F) and high level of clearance (Cl/F; Table 1) indicated a quick elimination of CL-55 from the blood and its distribution into the organs.

Time course of CL-55 (N-(2,4-difluorophenyl)-4-(3-ethoxy-4-hydroxybenzyl)-5-oxo-5,6-dihydro-4H-[1,3,4]-thiadiazine-2-carboxamide) plasma concentrations. The samples were collected at 5, 10, 15, 30, 60 and 120 min after administration of 2 mg CL-55 in 50 μl of polyethylene glycol-400. Samples from five mice were collected at each time point. A full color version of this figure is available at The Journal of Antibiotics journal online.
Discussion
In this study we showed for the first time that a heterocyclic compound from a group of thiohydrazides of oxamiс acids, CL-55, affects acute Salmonella infection in vivo. We demonstrated that CL-55 significantly decreased mortality and bacterial loads in susceptible and resistant mice infected with the S. enterica serovar Typhimurium. Injection of CL-55 at a dose of 10 mg kg−1 for 4 days significantly decreased the numbers of Salmonella in the spleens and resulted in a twofold increase in the survival rates in three strains of mice that differed in their resistance to Salmonella. Twelve days of therapy resulted in complete eradication of Salmonella in susceptible BALB/c mice. We also observed the absence of reactivation of infection during the additional two months post infection.
To address the activity of CL-55 on Salmonella infection in this study we employed the S. enterica serovar Typhimurium IV147 strain, which was found in our previous studies to possess a range of pathogenic features that makes it suitable for the aim of this study. First, this strain possesses sufficient virulence to induce an acute lethal infection with an i.p. LD100 dose of 2.5 × 102 CFU per mouse. This strain also induces lethal infection in classically susceptible (NRAMPS) BALB/c mice but also in I/St mice with intermediate susceptibility or in resistant A/Sn mice. In addition, this strain can induce chronic systemic infection via an oral route dose of 5 × 105 CFU per mouse in resistant A/Sn mice and in susceptible I/St mice (data not shown). The dynamics of survival and bacterial loads of the mice infected with this strain corresponds to those reported for other strains. This finding suggests that the data obtained in this study is representative for other Salmonella strains; however, additional studies are necessary to extend our finding to clinically important Salmonella strains.
The effect of T3SS inhibitors in vivo is described in a few studies. A decrease in the bacterial load and inflammation for Chlamydia, Salmonella and Yersinia but only as prophylactic treatment before infection17, 18, 19 was previously shown.
Eradication of C. rodentium in the model of colitis under the action of compound aurodox, therapy of which was started immediately after infection, was also reported;20 however, inhibiting the action of aurodox on T3SS was demonstrated for enteropathogenic E. coli but not for C. rodentium, and the mechanism of action of this compound on the course of C. rodentium infection is not clear. To the best of our knowledge, these are the first results that demonstrate a sufficient level of in vivo therapeutic potency from a compound with anti-T3SS activity. Previously, we found that CL-55, a compound with predicted anti-T3SS activity, displays specific activity toward T3SS and particularly decreased the translocation of IncA in the model of Chlamydia infection.21 Some preliminary data were also obtained regarding the activity of CL-55 toward SPI-1 and SPI-2 of Salmonella (unpublished data). In this study we focused on the therapeutic potential of CL-55 toward Salmonella infection; however, direct confirmation of its specificity toward T3SS of Salmonella needs further assessment.
Additionally, we demonstrated that CL-55 was not carcinogenic or mutagenic, did not increase the level of chromosomal aberrations in bone marrow cells and displayed low toxicity in mice, rats and rabbits.
Preclinical pharmacokinetics studies showed that CL-55 quickly accumulates and is retained in the organs with good blood supply, which may be due to the high lipophilic nature of this compound. Easy accumulation of the compound in the target organs suggests the possibility of achieving therapeutic effects with a relatively low level of the compound. The ability of the compound to be retained in the organs might be beneficial for the treatment of chronic infections. Our results demonstrate the potential of CL-55 for the development of new antibacterial drugs for the treatment of Salmonella infection.
References
Santos, R. L. Pathobiology of salmonella, intestinal microbiota, and the host innate immune response. Front. Immunol. 26, 252 (2014).
Chaand, M. et al. Type 3 secretion system island encoded proteins required for colonization by non-O1/non-O139 serogroup V. cholerae. Infect. Immun. 83, 2862–2869 (2015).
Kenny, B. Mechanism of action of EPEC type III effector molecules. Int. J. Med. Microbiol. 291, 469–477 (2002).
Marteyn, B., Gazi, A. & Sansonetti, P. Shigella: a model of virulence regulation in vivo. Gut Microbes 3, 104–120 (2012).
Wilharm, G. & Heider, C. Interrelationship between type three secretion system and metabolism in pathogenic bacteria. Front. Cell Infect. Microbiol. 27, 150 (2014).
Zigangirova, N. A., Nesterenko, L. N., Tiganova, I. G. & Kost, E. A. The role of the type-three secretion system of the gram-negative bacteria in regulation of chronic infections. Mol. Gen. Mikrobiol. Virusol. 3, 3–13 (2012a).
Knodler, L. A., Nair, V. & Steele-Mortimer, O. Quantitative assessment of cytosolic Salmonella in epithelial cells. PLoS ONE 9, e84681 (2014).
Marshall, N. C. & Finlay, B. B. Targeting the type III secretion system to treat bacterial infections. Expert. Opin. Ther. Targets 18, 137–152 (2014).
Gabrielsen, M. et al. Structural characterisation of Tpx from Yersinia pseudotuberculosis reveals insights into the binding of salicylidene acylhydrazide compounds. PLoS One 7, e32217 (2012).
Wang, D. et al. Identification of bacterial target proteins for the salicylidene acylhydrazide class of virulence-blocking compounds. J. Biol. Chem. 286, 29922–29931 (2011).
Chu, H. et al. Candidate vaginal microbicides with activity against Chlamydia trachomatis and Neisseria gonorrhoeae. Int. J. Antimicrob. Agents 36, 145–150 (2010).
Ur-Rehman, T. et al. Pre-clinical pharmacokinetics and anti-chlamydial activity of salicylidene acylhydrazide inhibitors of bacterial type III secretion. J. Antibiot. 65, 397–404 (2012).
Layton, A. N. et al. Salicylidene acylhydrazide-mediated inhibition of type III secretion system-1 in Salmonella enterica serovar Typhimurium is associated with iron restriction and can be reversed by free iron. FEMS Microbiol. Lett. 302, 114–122 (2010).
Keyser, P., Elofsson, M., Rosell, S. & Wolf-Watz, H. Virulence blockers as alternatives to antibiotics: type III secretion inhibitors against Gram-negative bacteria. J. Intern. Med. 264, 17–29 (2008).
Negrea, A. et al. Salicylidene acylhydrazides that affect type III protein secretion in Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 51, 2867–2876 (2007).
Veenendaal, A. K., Sundin, C. & Blocker, A. J. Small-molecule type III secretion system inhibitors block assembly of the Shigella type III secreton. J. Bacteriol. 191, 563–570 (2009).
Hudson, D. L. et al. Inhibition of type III secretion in Salmonella enterica serovar Typhimurium by small-molecule inhibitors. Antimicrob. Agents Chemother. 51, 2631–263 (2007).
Garrity-Ryan, L. K. et al. Small molecule inhibitors of LcrF, a Yersinia pseudotuberculosis transcription factor, attenuate virulence and limit infection in a murine pneumonia model. Infect. Immun. 78, 4683–4690 (2010).
Slepenkin, A., Chu, H., Elofsson, M, Keyser, P. & Peterson, E. M. Protection of mice from a Chlamydia trachomatis vaginal infection using a salicylidene acylhydrazide, a potential microbicide. J. Infect. Dis. 204, 1313–1320 (2011).
Kimura, M. et al. A small-molecule inhibitor of the bacterial type III secretion system protects against in vivo infection with Citrobacter rodentium. J. Antibiot. 64, 197–203 (2011).
Zigangirova, N. A. et al. Development of chlamydial type III secretion system inhibitors for suppression of acute and chronic forms of chlamydial infection. Acta Naturae 4, 87–97 (2012b).
Koroleva, E. A. et al. Small molecule inhibitor of type three secretion suppresses acute and chronic Chlamydia trachomatis infection in a novel urogenital Chlamydia model. Biomed. Res. Int. 2015, 484853 (2015).
Nesterenko, L. N. et al. Mycobacterium tuberculosis-susceptible I/St mice develop severe disease following infection with taxonomically distant bacteria, Salmonella enterica and Chlamydia pneumoniae. Clin. Exp. Immunol. 146, 93–100 (2006).
Schindewolf, C. et al. Comet assay as a tool to screen for mouse models with inherited radiation sensitivity. Mamm. Genome 11, 552–554 (2000).
Preston, R. J. et al. Mammalian in vivo cytogenetic assays. Analysis of chromosome aberrations in bone marrow cells. Mutat. Res. 189, 157–165 (1987).
Acknowledgements
This work was supported by grants from the Russian Ministry of Science and Education and the Russian Ministry of Industry.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on The Journal of Antibiotics website
Supplementary information
Rights and permissions
About this article

Cite this article
Nesterenko, L., Zigangirova, N., Zayakin, E. et al. A small-molecule compound belonging to a class of 2,4-disubstituted 1,3,4-thiadiazine-5-ones suppresses Salmonella infection in vivo. J Antibiot 69, 422–427 (2016). https://doi.org/10.1038/ja.2015.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2015.131
This article is cited by
-
Antibiotics in the clinical pipeline as of December 2022
The Journal of Antibiotics (2023)
-
A novel antivirulent compound fluorothiazinone inhibits Klebsiella pneumoniae biofilm in vitro and suppresses model pneumonia
The Journal of Antibiotics (2023)
-
Preventative treatment with Fluorothiazinon suppressed Acinetobacter baumannii-associated septicemia in mice
The Journal of Antibiotics (2022)
-
Fluorothiazinon, a small-molecular inhibitor of T3SS, suppresses salmonella oral infection in mice
The Journal of Antibiotics (2021)
-
Antibiotics in the clinical pipeline in October 2019
The Journal of Antibiotics (2020)
