
Abstract
Stem cells can modify macrophage phenotypes; however, the mechanisms remain unclear. We investigated whether n-butylidenephthalide (BP) primed adipose-derived stem cells (ADSCs) attenuated cardiac fibrosis via regulating macrophage phenotype by a PI3K/STAT3-dependent pathway in postinfarcted rats. Male Wistar rats after coronary ligation were allocated to receive either intramyocardial injection of vehicle, ADSCs (1 × 106 cells), BP-preconditioned ADSCs, (BP + lithium)-preconditioned ADSCs, (BP + LY294002)-preconditioned ADSCs, and (BP + S3I-201)-preconditioned ADSCs. ADSCs were primed for 16 h before implantation. BP-pretreated ADSCs increased the cell viability compared with naive ADSCs in the in vitro experiments. Infarct sizes were similar among the infarcted groups at the acute and chronic stages of infarction. At day 3 after infarction, post-infarction was associated with increased M1 macrophage infiltration, which was inhibited by administering naive ADSCs. Compared with naive ADSCs, BP-preconditioned ADSCs provided a significant increase of Akt and STAT3 phosphorylation, STAT3 activity, STAT3 nuclear translocation, myocardial IL-10 levels, and the percentage of M2 macrophage infiltration. The effects of BP on M2 polarization were reversed by LY294002 or S3I-201. Furthermore, the phosphorylation of both Akt and STAT3 was abolished by LY294002, whereas Akt phosphorylation was not affected following the inhibition of STAT3. The addition of lithium did not have additional effects compared with BP alone. After 4 weeks of implantation, ADSCs remained in the myocardium, and reduced fibrosis and improved cardiac function. BP-preconditioned ADSCs provided superior cardioprotection, greater ADSC engraftment, and antifibrotic effects compared with naive ADSCs. These results suggest that BP-pretreated ADSCs polarize macrophages into M2 cells more efficiently than naive ADSCs via the PI3K/STAT3 pathway.
Similar content being viewed by others

Introduction
Cardiomyocyte regeneration occurs after myocardial infarction (MI), however, it cannot prevent the injured area from being replaced by fibrous scars, which may contribute to the progression of heart failure. Fibrosis is modulated primarily by the inflammatory response at the site of injury. Among the cells involved in mediating inflammation, macrophages act as major effector cells in tissue repair and fibrosis [1]. Macrophages can be categorized as M1 and M2. M1 macrophages are often thought of as pro-inflammatory action and express inflammatory genes, including iNOS. In contrast, M2 macrophages are involved in the resolution of inflammation [2] and can secrete arginase-1 and IL-10 [3]. M2 macrophages can be further divided into 3 sub-populations: M2a, M2b, and M2c based on their characteristics and functions. The M2a phenotype is defined by high expression of arginase-1 and the M2c phenotype, IL-10 [3]. Excessive activation of M1 macrophages and delayed transition from M1 to M2 worsen post-MI remodeling [4]. Despite these recent advances, the signaling pathways by skewing M1 macrophages to the M2 phenotype are poorly understood. The signal transducer and activator of transcription 3 (STAT3) signaling pathway has been shown to be a key player regulating macrophage phenotype. STAT3 belongs to the STAT family of transcription factors that drives macrophage polarization to M2c phenotype. Very recently, sustained phosphatidylinositol 3-kinase (PI3K) activation has been shown to increase lung fibrosis by modulating the macrophage polarization [5]. Although PI3K and STAT proteins represent two distinct cellular regulatory systems, a functional link between these two systems has been shown by the observation that a specific inhibitor of PI3K reduces the level of STAT3 phosphorylation [6]. Thus, it is assumed that PI3K can tune macrophage phenotype at least in part by STAT3 translocation, however, the exact role of PI3K/STAT3 signaling on macrophage skewing remained unknown.
Adipose-derived stem cell (ADSC) transplantation has emerged as a promising new avenue for improving cardiac function after MI [7]. Mesenchymal stem cells are capable of reprogramming macrophages into M2 phenotype in vitro [8, 9] and in vivo [8, 10]. Transplantation of mesenchymal stem cells decreases M1 macrophage accumulation and increases the percentage of M2 macrophages in the infarcted hearts [11]. However, the specific molecular and cellular mechanisms that influence the skewing between the macrophage M1–M2 polarization by ADSCs remain unclear.
Although progress has been made in the selection of optimal transplantation conditions, little success has been achieved in the treatment efficiency of ADSC transplantation. Preconditioning of stem cells in culture is an emerging alternative therapeutic option to reduce the massive death of cells after transplantation. Cell therapy administered after infarct may mitigate the extent of injury by modulating macrophage infiltration and polarization is conceptually novel, a phenomenon called “cellular postconditioning”. Angelica sinensis, a traditional Chinese herbal medicine, has been used for the treatment of various health problems including cardiovascular diseases [12]. Butylidenephthalide (BP) is an active compound of Angelica sinensis [13]. BP has been reported to improve cognitive impairment via the activation of PI3K [14]. However, whether the effect of preconditioned ADSCs by BP on cardiac fibrosis was via activating PI3K signaling after infarction remains unclear. In this study, we investigated (1) whether in vivo transplantation of human ADSCs attenuated myocardial fibrosis through the regulation of macrophage phenotypes, (2) whether the anti-fibrotic effect of BP-pretreated ADSCs is superior to naive ADSCs, and (3) whether the role of PI3K and STAT3 is involved in cardiac fibrosis using inhibitors of PI3K and STAT3 in a rat MI model.
Methods
Male Wistar rats were purchased from LASCO (Taipei, Taiwan). All experiments were conducted according to protocols approved by the China Medical University Committee on Animal Care (Permit Number: 2016-070) and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Two to three rats were housed in temperature-controlled ventilated cabinets and monitored daily for any signs of distress or clinical symptoms of illness by trained personnel. At the end of the experiment, the rats were sacrificed under sodium pentobarbitone anesthesia according to the guidelines for euthanasia.
Isolation of human ADSCs
Human ADSCs were purchased from the commercially available kits (Stempro human ADSC kit; Invitrogen, Carlsbad, CA, USA) and were processed in an ISO17025 certified good manufacturing clinical grade Laboratory (Gwo Xi Stem Cell Applied Technology, Hsinchu, Taiwan) as previously described [15]. Initial establishment was deemed passage 1. ADSCs from passages 3–5 were used for experiments. These ADSCs were homogeneous without the markers of endothelial cells or hematopoietic lineages. Cultured ADSCs have been shown to display mesenchymal stem cell phenotype: they express the mesenchymal stem cell marker CD90 and do not express hematopoietic markers CD31 and CD45. They were confirmed to be >95% CD90+ and <2% CD31+/CD45+.
The cell viability of ADSCs was evaluated by analyzing DNA binding fluorophores. After treatment with different doses of n-BP (0, 7, 20 μg/ml) for 16 h, the cell numbers were assessed using the ADAM-MC™ Automatic Cell Counter (Digital Bio, NanoEnTek Inc., Seoul, Korea). The cells were mixed with AccuStain Solution T and N, respectively, and directly pipetted on to a disposable plastic chip. AccuStain Solution T for the total cell counting was composed of the fluorescent dye (propidium iodide, PI) and lysis solution. AccuStain Solution N for the non-viable cell counting was composed of the fluorescent dye and PBS. The viability was automatically calculated in the ADAM-MC software after each measurement of the total cells and the non-viable cells. A 532-nm optic laser was automatically focused on the cell suspension contained in a disposable microchip, and the cell analysis was conducted with a CDD camera.
Induction of MI and cell transplantation
The left anterior descending coronary artery of male Wistar rats (250–300 g) was ligated to induce MI under anesthesia with ketamine–xylazine (90 mg/kg–9 mg/kg, intraperitoneally) using a ventilator (Harvard Apparatus 486) as we previously described [16]. One hour after ligation, rats were randomized into groups of either vehicle or cell transplantation. For cell transplantation, ADSCs were detached from the plate, suspended in 30 μl of PBS (1 × 106 cells), and transplanted at three injection sites into the viable myocardium bordering the infarction using a syringe with a 30-gauge needle. ADSC were primed with either 7 μg/ml BP (Alfa Aesar), BP + 1 mM lithium (a PI3K agonist), BP + 20 µM LY294002 (a PI3K inhibitor), or BP + 25 µM S3I-201 (a STAT3 inhibitor; Calbiochem, La Jolla, CA, USA) for 16 h before transplantation. Lithium was used as a positive control as it is known that lithium pretreatment ameliorated lipopolysaccharide-induced microglial activation, by activating PI3K signaling [17]. The doses of BP [18], LiCl [19], LY294002 [20], and S3I-201 [21] were used as previously mentioned. Before cell transplantation, cells were washed with PBS 3 times to remove the direct drug or media effects. The heart tissues were collected at days 3 or 28 after MI as early and late stages of MI. The duration of the study was set at 4 weeks, as the myocardial remodeling process in rats has been reported to be mostly complete (70–80%) within 3 weeks [22]. Sham surgery served as the control group. Thus, together, the experimental groups included as follows: sham group and infarction groups (vehicle, ADSC, BP-ADSC, (BP/lithium)-ADSC, (BP/LY294002)-ADSC, and (BP/S3I-201)-ADSC).
Echocardiogram
At 28 days after operation, the rats after intraperitoneal administration of ketamine–xylazine (45 mg/kg–5 mg/kg) underwent echocardiographic measurements as shown in the Supplementary material online.
Hemodynamics and infarct size measurements
Hemodynamic parameters and infarct size were measured in anesthetized rats at the end of the study available as online Supplementary material.
Western blot analysis of Akt, STAT3, iNOS, IL-10, and α-smooth muscle actin (α-SMA)
Protein levels were quantified by using Western blot, with β-actin as a loading control. For detailed methods, please refer to Supplementary Methods.
Real-time RT-PCR of IL-6, IL-1β, iNOS, CD206, and IL-10
mRNAs were quantified by real-time quantitative reverse transcription-polymerase chain reaction (RT-PCR) using TaqMan system (Prism 7700 Sequence Detection System, PE Biosystems) with cyclophilin as a housekeeping gene at day 3 after MI. We measured the gene expression patterns for M1 (IL-6, IL-1β, iNOS) and M2 (CD206, IL-10) macrophages. The sequence of PCR primers is shown in the online data supplement.
Immunohistochemical analysis of Akt, STAT3, CD68, iNOS, IL-10, α-SMA, human mitochondria, and sarcomeric α-actinin
To confirm the downstream pathways of PI3K/Akt signaling, immunohistochemical staining was performed on LV muscle 3 days for M1 and M2 analysis and 28 days for myofibroblast and engrafted ADSCs. Myocardial cryosections were incubated with antibodies against phosphor(ser473)-Akt1 (Cell Signaling Technology, Beverly, MA, USA), phospho(Tyr705)-STAT3 (Cell Signaling Technology, Danvers, MA, USA), CD68 (a marker for all macrophages; Abcam, Cambridge, MA, USA), iNOS (a marker for M1; Cell Signaling Technology, Danvers, MA, USA), IL-10 (a marker for M2c; R&D Systems, Abingdon, UK), and α-SMA (a marker for myofibroblast; Sigma, St. Louis, MO, USA) as online Supplementary material.
Morphometry of cardiac fibrosis
Heart sections from the remote regions were stained with Sirius red stain to measure interstitial fibrosis. For detailed methods, please refer to Supplementary Methods.
Laboratory measurements
The culture supernatants from ADSCs were collected after being treated for 16 h. The concentrations of paracrine factors, including insulin-like growth factor 1 (IGF-1) and hepatocyte growth factor (HGF), were measured by ELISA (Sigma-Aldrich Corp., St. Louis, MO, USA) kits according to the manufacturer’s instructions.
The samples from the remote zone were frozen immediately upon collection in liquid nitrogen and stored at −80 °C until assayed. To measure the DNA-binding activity of STAT3, a TransAM STAT3 Transcription Factor Assay Kit (Active Motif) was used to assess the activity in myocardial homogenates according to the manufacturer’s protocol. Histological collagen analyses were further determined by the hydroxyproline assay according to Stegemann and Stalder [23]. The results were expressed as hydroxyproline content per weight of tissue. Each experiment was carried out as duplicate and was repeated for three times.
To measure myocardial IL-10 activity as a biomarker of M2c, we detected myocardial membrane-bound IL-10 fractions using commercially available ELISA kits (R&D Systems) in myocardial homogenates from the border zones. For detailed methods, please refer to Supplementary Methods.
Statistical analysis
All data are expressed as ±standard deviation (SD). Statistical analysis was conducted with the SPSS statistical package (SPSS, version 19.0, Chicago, IL, USA). One-way ANOVA was used to determine differences among the groups. Bonferroni’s correction was performed if significant differences between the two groups were observed. A P value of <0.05 was considered to indicate statistical significance.
Results
In vitro studies
Effect of BP-pretreated ADSCs on cell viability
An automated cell counter assay was performed to detect the effect of BP at different doses on the viability of ADSCs. ADSCs pretreated with BP at 7 μg/ml demonstrated viability significantly higher than that in naive ADSCs (94.5 ± 1.7% in BP-treated ADSCs vs. 92.0 ± 2.3% in naive ADSCs, P < 0.05) (Fig. 1a). However, there was no additional effect of BP administration at a higher concentration of 20 μg/ml on the viability of ADSCs.

In vitro measurements. a Cell viability. b ELISA detection of IGF-1 and HGF. Each column and bar represents mean ± SD. N = 7–8 for each group. *P < 0.05 compared with naive ADSCs; †P < 0.05 compared with naive ADSCs-, ADSC/BP/LY294002-, and ADSC/BP/S3I-201-treated infarcted groups; ‡P < 0.05 compared with ADSC/BP-treated group
Effect of BP-pretreatment of ADSCs on soluble factors
ELISA assays were performed to evaluate the effect of BP on cytokine secretion in ADSCs. As shown in Fig. 1b, the levels of IGF-1 (54.6 ± 2.2 pg/ml in BP-treated ADSCs vs. 50.7 ± 2.6 pg/ml in naive ADSCs, P < 0.05) and HGF (48.4 ± 3.2 pg/ml in BP-treated ADSCs vs. 22.4 ± 1.8 pg/ml in naive ADSCs, P < 0.05) were significantly increased after BP treatment. The effect was abolished by the addition of LY294002 and S3I-201.
Acute stage (day 3)
Heart tissue was collected at day 3 after MI. There were no significant differences in mortality and infarct size among the infarcted groups at the acute stage of MI.
Effect of BP-pretreated ADSCs on the engraftment
The quantification of the human origin of these transplanted ADSCs was performed by staining with anti-human mitochondria antibody 3 days after transplantation (Fig. 2). As expected, human stem cells were found in the implanted area of the transplantation group (arrows) but not the vehicle group. In the transplantation hearts, a small number of mitochondria-positive human cells were observed in clusters, mainly at the border zone. The mitochondria-positive cells were observed more frequently in the rats administered BP. The ratio of mitochondria-positive cells in the BP-primed ADSC group was 0.93 ± 0.25%, significantly higher than in the naive ADSC group (0.39 ± 0.12%, P < 0.05). These data showed that BP administration improved the survival rate of ADSCs in post-infarct hearts.

Identification of the transplanted human ADSCs and their cardiac phenotype 3 days after infarction. The grafted cells identified by their human nature were confirmed by positive immunostaining with anti-human mitochondrial antibodies (brown). Human stem cells were observed at the implanted area of transplantation groups but not the vehicle. More grafted cells were found in the infarcted group pretreated with BP compared with naive ADSCs. N = 7–8 for each group. *P < 0.05 compared with naive ADSCs
Effect of BP-pretreated ADSC transplantation on Akt and STAT3 activation
Western blotting showed that the phosphorylation of Akt and STAT3 (p-tyr705) was significantly increased after MI at 3 days (P < 0.05 vs. sham, Fig. 3a). Compared with vehicle alone, STAT3 activation was significantly enhanced to 157 ± 9% of vehicle (P < 0.001) after ADSC implantation. BP-primed ADSCs had a further increase in relative p-Akt levels and p-STAT3 levels compared with those in naive ADSCs (both P < 0.05). LY294002, the PI3K inhibitor, not only eliminated Akt activation but also inhibited STAT3 phosphorylation to a similar extent as with vehicle. However, the STAT3-specific inhibitor, S3I201, decreased only the phosphorylation of STAT3, but did not affect Akt phosphorylation. These results suggest that Akt likely serves as an upstream molecule of STAT3.

Akt and STAT3 activity analysis from the border zone at day 3 after MI. a Western analysis of Akt and STAT3. Relative abundance was obtained by normalizing the protein density against that of β-actin. Each column and bar represents mean ± SD. Each experiment was repeated for three times in duplicates. b DNA-binding activity of STAT3 was measured by ELISA with heart homogenates. c Representative immunostaining micrographs show STAT3 nuclear translocation (brown). BP increased the nuclear translocation of STAT3 in infarcted hearts. The value was expressed as the ratio of STAT3-stained area to total area. Bar = 50 μm. d To identify the cell types of expressing Akt and STAT3, double staining with CD68 and signaling markers was performed. BP-pretreated ADSCs showed a significant increase of Akt and STAT3 phosphorylation in cardiac macrophages. The p-Akt1-expressing or p-STAT3-expressing macrophages were calculated and expressed as bar graphs. Bar = 20 μm. N = 7–8 for each group. *P < 0.05 compared with sham; †P < 0.05 compared with vehicle- and ADSC/BP/LY294002-treated infarcted groups; ‡P < 0.05 compared with ADSC-treated group; §P < 0.05 compared with ADSC/BP-treated group; #P < 0.05 compared with vehicle-treated infarcted group
The DNA-binding activity of STAT3 was consistently significantly increased in BP-pretreated ADSC infarcted rats compared with naive ADSC-treated rats, as measured by transcription factor ELISA (Fig. 3b).
To further confirm the activation of myocardial STAT3, immune-staining analysis with anti-p-tyr705 STAT3 antibody was performed to evaluate the nuclear translocation (Fig. 3c). As a result, myocardial p-STAT3 can be seen in the nucleus after MI. The extent of STAT3 nuclear translocation was significantly increased in ADSC-treated infarcted rats than in the vehicle-treated infarcted rats. As expected, the nuclear localization of p-STAT3 was dramatically enhanced by BP administration.
To evaluate the expression of p-Akt and p-STAT3 in cardiac macrophages, sections were stained using antibodies specific for macrophage markers and signaling markers. Akt1 activation was significantly increased in cardiac macrophages in the BP-pretreated group compared with naive ADSCs (26.3 ± 6.5% in BP-treated ADSCs vs. 15.2 ± 4.2% in the naive ADSCs, P < 0.05, Fig. 3d). Similarly, nuclear phospho-STAT3-positive cells were significantly increased in cardiac macrophages in the BP-pretreated group compared with naive ADSCs (P < 0.05).
Effect of BP-pretreated ADSC transplantation on macrophages skewing toward an M2 phenotype through STAT3 signaling
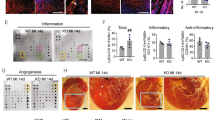
To explore the interactions of human ADSCs and macrophage differentiation at the wounds, type-specific surface markers of macrophages were assessed. The markers for M1 (CD68+, iNOS+) and M2c (CD68+, IL-10+) macrophages were examined in infarcted myocardium (Fig. 4). Immunohistochemical staining revealed that CD68+ macrophages were infiltrated in the infarcted groups at day 3 after MI. Compared with vehicle, iNOS-expressing CD68+ macrophages were significantly decreased in the naive ADSCs (18.5 ± 5.8% in the vehicle vs. 10.4 ± 4.2% in the naive ADSCs, P < 0.05, Fig. 4a), and IL-10-expressing CD68+ macrophages were more frequent in the naive ADSCs (4.3 ± 3.8% in the vehicle vs. 7.5 ± 4.2% in the naive ADSCs, P < 0.05, Fig. 4b). Significantly, BP-primed ADSCs decreased the percentage of M1 macrophages by 77% and increased the percentage of M2 macrophages by 133% 3 days after MI, compared with naive ADSCs (P < 0.01). These data suggest improved ability of BP-primed ADSCs-treated rats to increase M2 macrophage differentiation with a concomitant reduction in M1.

M1 and M2 macrophage analysis by immunohistochemical staining, gene marker expression, and function at the border zone at day 3 after MI. a iNOS-expressing CD68 (+) M1 macrophages were recruited in infarcted myocardium treated with vehicle (Veh), but were significantly reduced by either ADSC or ADCS/BP administration. b IL-10-expressing CD68 (+) M2 macrophages were predominant in BP-administered infarcted myocardium. The iNOS-expressing CD68 (+) or IL-10-expressing CD68 (+) macrophages were calculated and expressed as bar graphs. c mRNA, d protein levels, e myocardial IL-10 activity, and f myocardial arginase activity. There is an obvious shift towards M2 macrophage phenotype in the infarcted group pretreated with BP as shown by changed expression of reduced M1-related genes (IL-6, IL-1β, iNOS) and increased M2 genes (CD206, IL-10). N = 7–8 for each group. The line length corresponds to 20 μm. M: merged. *P < 0.05 compared with vehicle; †P < 0.05 compared with vehicle- and ADSC-treated infarcted groups; ‡P < 0.05 compared with vehicle-, ADSC/BP/LY294002-, and ADSC/BP/S3I-201-treated infarcted groups; #P < 0.05 compared with vehicle-, ADSC-, ADSC/BP/LY294002-, and ADSC/BP/S3I-201-treated infarcted groups
To investigate whether the STAT3 signaling pathway is critical for the polarization of M2c macrophages, we examined the effect of S3I-201 on macrophage transformation into M2c by RT-PCR, Western blot, and ELISA. The expression of M1 mRNA (IL-6, IL-1β, iNOS) was remarkably decreased and the expression of M2 mRNA (CD206, IL-10) was highly induced in the infarcted groups treated with ADSCs and BP-primed ADSCs (Fig. 4c). The extent of M2 mRNA was significantly higher in the BP-primed ADSCs than that in the naive ADSCs. Furthermore, the expression of M1 mRNA and M2 mRNA was not significantly changed after adding lithium compared with BP-primed ADSCs. However, the increased M2 gene expression in the BP-primed ADSC group was reduced after administering either LY294002 or S3I-201. Then, Western blotting of iNOS and IL-10 protein levels was performed. The IL-10 levels markedly increased in the BP-primed ADSC group. However, the increased IL-10 levels in the BP-primed ADSCs group was reduced after administering S3I-201. The protein ratio of iNOS to IL-10 was calculated as shown in Fig. 4d. These results show that BP-primed ADSCs lead to inhibitory responses on M1 markers and stimulatory effects on M2 markers.
Figure 4e shows myocardial IL-10 activity measured by ELISA. IL-10 activity levels were significantly higher (P < 0.05) in the BP-primed ADSCs than in naive ADSCs. However, the IL-10 activity was significantly decreased after adding S3I-201. There was similar total arginase activity among the infarcted groups (Fig. 4f).
Chronic stage (day 28)
There was similar mortality among the infarcted groups during 4 weeks after infarction. (Table 1). Four weeks after infarction, the anterior wall of the infarcted LV was replaced completely by a thin layer of scar tissue. Relative LV weights normalized by body weight at the end of the experimental period are not significant differences among groups, except the lithium treatment group. The ratios of right ventricular weight/body weight and lung weight/body weight, indexes of lung edema, were significantly lower in the ADSC-treated infarcted group than in the vehicle-treated infarcted group. The ratios were significantly further lower in the ADSC/BP- and ADSC/BP/Li-treated infarcted groups compared with those in the naive ADSC group. The beneficial effect of BP on the ratios of right ventricular weight/body weight and lung weight/body weight was abolished after adding either LY294002 or S3I-201. The values of +dp/dt and −dp/dt were significantly higher in the ADSC/BP- and ADSC/BP/Li-treated infarcted groups compared with those in the ADSCs, ADSCs/BP/LY294002 and ADSCs/BP/S3I-201. There were no significant differences in LV end-systolic pressure, LV end-diastolic pressure, and infarct size among the infarcted groups.
Effect of BP-pretreated ADSCs on the long-term transdifferentiation
To assess transdifferentiation of human ADSC-derived ventricular cardiomyocytes, the coexpression of human mitochondria and sarcomeric α-actinin antibodies was used for confirmation of donor-derived cardiomyocyte phenotype. In small engrafted cells, colocalization of human cell marker and cardiomyocyte-specific marker was identified (Fig. 5). Compared with naive ADSCs, BP-primed ADSCs showed a higher transdifferentiation rate (0.21 ± 0.10% vs. 0.11 ± 0.08% in naive ADSCs, P < 0.05).

Identification of cardiac phenotype of the transplanted ADSCs 28 days after infarction. Cardiomyocyte phenotype of transplanted human ADSCs was confirmed by coexpression of human mitochondria (green) and sarcomeric α-actinin (cardiomyocyte phenotype, red) in an infarcted rat treated with BP-pretreated ADSCs. In the merged (M) image, coexpression of human mitochondria and sarcomeric α-actinin showed colocalization of antigen expression as indicated by yellow fluorescence. N = 7–8 for each group. *P < 0.05 compared with naive ADSCs
Effects of BP-pretreated ADSC transplantation on cardiac function
To investigate whether BP enhanced the therapeutic function of stem cells, cardiac function was assessed by echocardiography (Table 1). LV ejection fraction after MI was significantly reduced. Ejection fraction was significantly improved in all ADSC-treatment groups compared with vehicle. Although there were similar LV ejection fractions among all ADSCs groups, the LVESD and LVEDD in the groups treated with ADSC/BP or ADSC/BP/Li were significantly reduced compared with those in ADSCs, ADSC/BP/LY294002 and ADSC/BP/S3I-201.
Effects of BP-pretreated ADSCs on myofibroblasts by immunohistochemical staining and Western blots
α-SMA-expressing myofibroblasts appeared in the interstitium of the infarcted myocardium 4 weeks after infarction (Fig. 6a). The α-SMA signals significantly increased in vehicle-treated rats than in sham. Either ADSC- or BP-primed ADSC administration significantly reduced the intensity of the immunoreaction compared with respective vehicles. To further confirm the findings of immunohistochemical staining, Western blots of α-SMA were performed. Similarly, the protein levels of α-SMA were significantly reduced in the ADSC- and ADSC/BP-treated groups compared with that in the vehicle (Fig. 6b). However, the attenuation of α-SMA levels in the ADSC/BP group was reversed by the addition of either LY294002 or S3I-201.

Myofibroblast infiltration and cardiac fibrosis at the remote zone at day 28 after infarction. a Representative immunohistochemical staining of α-SMA, a hallmark of myofibroblasts (magnification 400×). b To further confirm the protein levels of α-SMA, Western blot was performed. Western blotting is consistent with the results of immunohistochemical staining. c Representative sections from the remote zone with Sirius Red staining (red, magnification 400×). d Hydroxyproline content to measure quantitative amount of fibrosis. The line length corresponds to 50 μm. N = 7–8 for each group. *P < 0.05 compared with sham, vehicle-, ADSC/BP/LY294002-, and ADSC/BP/S3I-201-treated infarcted groups; †P < 0.05 compared with ADSC-treated infarcted group
Effects of BP-pretreated ADSCs on remote myocardial fibrosis
Sirius Red staining to demonstrate fibrosis was examined in tissue sections of the LV from the remote zone as shown in Fig. 6c. The vehicle-treated infarcted rats had significantly larger areas of intense focal fibrosis compared with sham-operated rats (5.61 ± 0.91% vs. 0.04 ± 0.02%, P < 0.0001). Compared with vehicle-treated infarcted rats, treatment with naive ADSCs attenuated fibrosis. Compared with naive ADSCs, treatment with BP-pretreated ADSCs further attenuated fibrosis. Fibrosis was significantly increased in rats treated with a combination of BP-pretreated ADSCs and S3I-201 compared with BP-pretreated ADSCs-treated rats alone.
The hydroxyproline assay consistently showed a significant increase in collagen contents after MI, and this was significantly attenuated by ADSC administration (Fig. 6d).
Discussion
In this study, we show that transplantation of BP-preconditioned ADSCs attenuated cardiac fibrosis after MI, most likely through activation of PI3K/STA3-dependent M2 macrophages. Our results confirm and extend the findings of Vasandan et al. [24], which showed a M1 to M2 shift in inflammatory macrophages upon mesenchymal stem cell priming. These results are consistent with the beneficial effects of BP, as documented structurally by a reduction in cardiac fibrosis, improved engraftment, increased STAT3 nuclear translocation and M2 infiltration, and attenuated myofibroblast infiltration; molecularly by myocardial Akt, STAT3, iNOS and IL-10 protein and mRNA levels; biochemically by STAT3 DNA binding activity, IL-10 levels, and tissue hydroxyproline levels; pharmacologically by agonists and antagonists; and functionally by improvement of cardiac contractility. To the best of our knowledge, this finding identifies a previously unreported role of PI3K/STAT3 as a molecular link between BP mediating macrophage phenotypes with therapeutic potential after infarction. Indeed, our results are in accordance with the findings of Wang et al. [25], showing that the activation of the PI3K/Akt signaling polarizes macrophages toward M2. Furthermore, our results are consistent with the previous studies, showing that cardiac fibrosis was exaggerated in STAT3-deficient hearts after MI [26] and delivery of a human STAT3 gene reduced macrophage infiltration [27].
ADSCs may produce and release local signaling molecules that limit local inflammation when injected into injured tissues. BP-pretreated ADSCs promote wound healing without fibrosis than naive ADSCs. PI3K/STAT3 signaling is an essential pathway for the regulation of macrophage polarization and wound healing after MI. Our conclusions are supported by 3 lines of evidence (Fig. 7).

Reaction sequences illustrate the involvement of BP-pretreated ADSCs in macrophage skewing- and myofibroblast-mediated cardiac fibrosis after MI. Inflammatory M1 macrophages were dominant at the early stage of MI, however, anti-inflammatory M2 phenotype was prevalent at a late stage. IL-10 activity in M2 macrophages inhibits myofibroblast differentiation, which in turn reduces extracellular matrix production. Administration with BP-pretreated ADSCs exhibits potent antifibrotic activity via increasing the activation of M2 macrophages and inhibiting myofibroblast differentiation by activating PI3K/STAT3 pathway. Inhibition of these signaling pathways by their respective inhibitors is indicated by the vertical lines
BP increased phosphorylation of Akt
We noted increased Akt activity after MI. To further test the role of PI3K/Akt signaling in BP-pretreated ADSCs, we used LY294002 to block PI3K downstream signaling. This treatment abolished the BP-induced increase of p-Akt. Furthermore, the PI3K activator, lithium, had no additional effects, possibly because both BP and ADSCs already increased PI3K activity maximally. Thus, our study is consistent with the findings of Chen et al. [28], showing BP-induced tumor growth inhibition by activation of PI3K/Akt-dependent pathway.
Our results show that BP improved ADSC engraftment and transdifferentiation. The results are consistent with the notion that the PI3K/Akt signaling pathway is crucial for stem cell survival [29]. The survival of engrafted ADSCs was confirmed by the finding of anti-human mitochondrial antibody-positive tissue in the myocardium 4 weeks after cell transplantation. The engrafted ADSCs had further differentiated into cardiac tissue, as reflected by positive staining to cardiac-specific sarcomeric α-actinin. The engraftment and transdifferentiation were markedly improved in BP-preconditioned ADSCs compared with naive ADSCs. The in vivo study is consistent with the in vitro findings, showing that BP has a protective effect on the proliferation of ADSCs.
Akt phosphorylation modulated STAT3 activity
We show that there is cross-talk between the PI3K/Akt and STAT3 pathways. The PI3K pathway connects to many other signaling axes to form networks. However, it remains unclear whether there was crosstalk between PI3K and STAT3 signaling. PI3K and Stat proteins represent two distinct signaling systems that are not known to share the same molecules. We observed that blocking the PI3K pathway with LY294002 abolished the phosphorylation of both Akt and STAT3, whereas Akt activity was not eliminated following the inhibition of STAT3. These results indicate that Akt serves as an upstream molecule of STAT3 in the setting of BP-primed ADSCs. Our results are consistent with the findings of Shi et al. [30], showing that LY294002 down-regulated Akt and STAT3 phosphorylation and there was a crosstalk between their associated signaling cascades.
STAT3 activity is a key step for regulating macrophage polarization
MI increased the abundance of STAT3 phosphorylation at tyrosine 705. Therapy with either ADSCs or BP-preconditioned ADSCs has been shown to further enhance the STAT3 activity. BP-preconditioned ADSCs enhanced M2 macrophage infiltration and decreased myofibroblast infiltration at the border zone, which can be abolished by the addition of S3I-201, confirming the role of STAT3 in regulating macrophage-mediated fibroblast-to-myofibroblast differentiation.
M2 macrophages play an essential role in the control of inflammatory processes through the release of IL-10 and arginase [3]. As shown in Fig. 4b, IL-10 is highly expressed in the macrophages in the rats pretreated with ADSC. Our recent study has shown that infarcted hearts treated with anti-IL-10 receptor antibody may have significant activation of myofibroblasts [31]. When M2 polarization is directly inhibited by anti-IL-10 receptor antibody, the fibroblast activation is reduced. The role of IL-10 in macrophages modulating fibroblast activation and fibroregulation is consistent with previous studies, showing that scar formation was increased during wound healing in fetal mice by knocking out IL-10 [32], and then over-expression of IL-10 lead to scarless healing [33].
Additionally, our in vitro results show that BP-pretreated ADSCs directly increase paracrine secretion of IGF-1 and HGF, which may contribute to the inhibition of cardiac fibrosis. In the present study, BP was revealed to be an upstream activator of PI3K, and PI3K inhibition abolished the cytokine effects of BP. Our results are consistent with previous studies, showing that mesenchymal stem cells directly secrete a variety of cytokines and growth factors that have anti-fibrotic properties, including HGF [34]. Furthermore, HGF can resist myofibroblastic differentiation [35]. These combined mechanisms enable ADSCs to regulate the cells infiltrating the wound to favor healing to produce less scar. However, before cell transplantation, cells were washed with PBS 3 times to remove the paracrine effects. Although we cannot exclude the paracrine effect of ADSCs on cardiac fibrosis, the effect may be minimal.
Other mechanisms
Although our results provide evidence that the mechanisms of BP-primed ADSC’s attenuation of cardiac fibrosis may be related to increased STAT3-dependent M2 phenotype, other potential mechanisms need to be studied, such as antioxidant effect and cytokine-mediated effects. Reactive oxygen species have been shown to intensify collagen deposition [36]. Given nitric oxide produced by mesenchymal stem cells in the wound can scavenge reactive oxygen species to produce reactive nitrogen species, such as peroxynitrite [37]. It is not surprising to know that ADSCs attenuated cardiac fibrosis via a reactive oxygen species-dependent pathway.
Clinical implications
Despite the deleterious consequences of fibrosis and scar tissue formation, current treatments to promote wound healing without fibrosis are very limited. Of most significance was our finding that its strategy can be achieved by modulating macrophage phenotype. Manipulation of macrophage plasticity has been shown to be one of the underlying processes behind the miraculous ADSC-mediated reparative benefits after MI. The immunomodulatory effects of ADSCs on macrophage functions during wound repair suggest that therapeutic strategies can be developed for anti-fibrosis that more fully exploit the plasticity of ADSCs by driving their macrophage features in a reparative direction. Shifting macrophages from M1 into M2 phenotype can promote the wound repair, especially in the chronic wounds to minimize the visual appearance of scar tissue. Furthermore, BP-primed ADSCs showed superior beneficial effects on attenuated cardiac fibrosis compared with naive ADSCs. Besides, although myofibroblasts are crucial for proper wound repair, the need for their timely disappearance is important, otherwise fibrotic complications can occur [38]. Myofibroblasts have been shown to induce slow conduction and worsen ectopic activity in the presence of heterocellular electrotonic coupling with cardiomyocytes [39]. Furthermore, myofibroblasts isolated from MI rat hearts have been shown to exhibit significantly higher nerve growth factor expression [40], which in turn increased sympathetic nerve sprouting and worsened arrhythmias [16]. The present study explored the possibility that BP might have clinical efficacy for attenuated arrhythmias of ADSCs by attenuated myofibroblast infiltration after MI.
Study limitations
There were several limitations to this study. First, MI-induced rat death occurred mainly during the first 3 days after surgery, which was not significantly affected by BP-preconditioned ADSC transplantation. The lack of the effect on animal mortality suggests that although BP induced better ADSCs survival in the ischemic heart, the effect was not adequate to antagonize the initial overwhelming insults at the acute phase of MI. Moreover, the BP-preconditioned ADSC cell survival is expected to primarily benefit long-term tissue repair and functional improvement many days after MI, as shown in this study. Second, given that diastolic dysfunction is highly correlated with cardiac fibrosis [41], the measurement of diastolic function is a clinical outcome of cardiac fibrosis. However, except for −dp/dt, other physiological parameters for monitoring LV diastolic function including pressure–volume loop and diastolic radial and circumferential strain rates, were not provided in the current study. Indeed, our study may bridge the gap between macrophage phenotypes and diastolic dysfunction [42]. Third, STAT3 is a master transcriptional regulator of immune responses [43]. Although the hADSC-enhanced STAT3-dependent increase in IL-10 expression has been proposed to be one of the mechanisms for the phenotypic conversion of macrophages, the molecular mechanism of STAT3 in the macrophages polarization conversion was not explored in this study. The molecular signaling pathway by which STAT3 is involved in macrophage-induced IL-10 expression has been previously demonstrated. Sun et al. [44] have shown that increased STAT3 expression contributes to the negative regulation of dendritic cell function via enhancing the IL-10 signaling pathway. Furthermore, STAT3’s transcriptional program during the anti-inflammatory response has been shown to be highly specific to IL-10–stimulated M2 macrophages [45]. Recently, IL-10 locus has been shown to be one of the STAT3 binding peaks in the proximal promoter regions, implying that IL-10 is the downstream target of STAT3 [46]. Indeed, our results are consistent with the findings of Hutchins et al. [45], showing that IL-10 is one of the genomic targets of STAT3 during the anti-inflammatory response in macrophages. Finally, all study results were obtained by pharmacological inhibition. Pharmacological inhibitors may have many potential off-target effects. It would be of interest to further evaluate the in vivo antifibrotic effects of BP and ADSCs by using STAT3 knockout and transgenic animal models.
Conclusions
These data provide new evidence that pretreatment of ADSCs from xenogeneic donors with BP polarized macrophages towards M2 phenotype and attenuated cardiac fibrosis via activation of the PI3K/STAT3 pathway. The study provides novel mechanisms of BP-preconditioned ADSCs as a potential antifibrogenic modality in the prevention and treatment of cardiac fibrosis after MI, which has clinical consequences of particular significance.
References
Matsushita K, Dzau VJ. Mesenchymal stem cells in obesity: insights for translational applications. Lab Invest. 2017;97:1158–66.
Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86.
Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik DE, Aikawa E, et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55:1629–38.
Hart JR, Liao L, Yates JR 3rd, Vogt PK. Essential role of Stat3 in PI3K-induced oncogenic transformation. Proc Natl Acad Sci USA. 2011;108:13247–52.
Kral JB, Kuttke M, Schrottmaier WC, Birnecker B, Warszawska J, Wernig C, et al. Sustained PI3K activation exacerbates BLM-induced lung fibrosis via activation of pro-inflammatory and pro-fibrotic pathways. Sci Rep. 2016;6:23034.
Bai X, Yan Y, Song YH, Seidensticker M, Rabinovich B, Metzele R, et al. Both cultured and freshly isolated adipose tissue-derived stem cells enhance cardiac function after acute myocardial infarction. Eur Heart J. 2010;31:489–501.
Abumaree MH, Al Jumah MA, Kalionis B, Jawdat D, Al Khaldi A, Abomaray FM, et al. Human placental mesenchymal stem cells (pMSCs) play a role as immune suppressive cells by shifting macrophage differentiation from inflammatory M1 to anti-inflammatory M2 macrophages. Stem Cell Rev. 2013;9:620–41.
Maggini J, Mirkin G, Bognanni I, Holmberg J, Piazzón IM, Nepomnaschy I, et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS ONE. 2010;5:e9252.
Dayan V, Yannarelli G, Billia F, Filomeno P, Wang XH, Davies JE, et al. Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic Res Cardiol. 2011;106:1299–310.
Ben-Mordechai T, Holbova R, Landa-Rouben N, Harel-Adar T, Feinberg MS, Abd Elrahman I, et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. 2013;62:1890–901.
Yi LZ, Liang YZ, Wu H, Yuan DL. The analysis of Radix Angelicae sinensis (Danggui). J Chromatogr A. 2009;1216:1991–2001.
Ying L, Si-Wang W, Hong-Hai T, Wei C. Simultaneous quantification of six main active constituents in Chinese Angelica by high-performance liquid chromatography with photodiode array detector. Pharmacogn Mag. 2013;9:114–9.
Xiang J, Pan J, Chen F, Zheng L, Chen Y, Zhang S, et al. L-3-n-butylphthalide improves cognitive impairment of APP/PS1 mice by BDNF/TrkB/PI3K/AKT pathway. Int J Clin Exp Med. 2014;7:1706–13.
Lee TM, Harn HJ, Chiou TW, Chuang MH, Chen CH, Lin PC, et al. Targeting the pathway of GSK-3β/nerve growth factor to attenuate post-infarction arrhythmias by preconditioned adipose-derived stem cells. J Mol Cell Cardiol. 2017;104:17–30.
Lee TM, Lai PY, Chang NC. Effect of N-acetylcysteine on sympathetic hyperinnervation in post-infarcted rat hearts. Cardiovasc Res. 2010;85:137–46.
Dong H, Zhang X, Dai X, Lu S, Gui B, Jin W, et al. Lithium ameliorates lipopolysaccharide-induced microglial activation via inhibition of toll-like receptor 4 expression by activating the PI3K/Akt/FoxO1 pathway. J Neuroinflamm. 2014;11:140.
Harn HJ, Lin SZ, Lin PC, Liu CY, Liu PY, Chang LF, et al. Local interstitial delivery of z-butylidenephthalide by polymer wafers against malignant human gliomas. Neuro Oncol. 2011;13:635–48.
Nemoto T, Kanai T, Yanagita T, Satoh S, Maruta T, Yoshikawa N, et al. Regulation of Akt mRNA and protein levels by glycogen synthase kinase-3beta in adrenal chromaffin cells: effects of LiCl and SB216763. Eur J Pharmacol. 2008;586:82–9.
Abdul-Ghani R, Serra V, Györffy B, Jürchott K, Solf A, Dietel M, et al. The PI3K inhibitor LY294002 blocks drug export from resistant colon carcinoma cells overexpressing MRP1. Oncogene. 2006;25:1743–52.
Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, et al. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 2010;78:257–68.
Bélichard P, Savard P, Cardinal R, Nadeau R, Gosselin H, Paradis P, et al. Markedly different effects on ventricular remodeling result in a decrease in inducibility of ventricular arrhythmias. J Am Coll Cardiol. 1994;23:505–13.
Stegemann H, Stalder K. Determination of hydroxyproline. Clin Chim Acta. 1967;18:267–73.
Vasandan AB, Jahnavi S, Shashank C, Prasad P, Kumar A, Prasanna SJ. Human mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci Rep. 2016;6:38308.
Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci USA. 2015;112:2853–8.
Enomoto D, Obana M, Miyawaki A, Maeda M, Nakayama H, Fujio Y. Cardiac-specific ablation of the STAT3 gene in the subacute phase of myocardial infarction exacerbated cardiac remodeling. Am J Physiol Heart Circ Physiol. 2015;309:H1471–80.
Khan JA, Cao M, Kang BY, Liu Y, Mehta JL, Hermonat PL. AAV/hSTAT3-gene delivery lowers aortic inflammatory cell infiltration in LDLR KO mice on high cholesterol. Atherosclerosis. 2010;213:59–66.
Chen YL, Jian MH, Lin CC, Kang JC, Chen SP, Lin PC, et al. The induction of orphan nuclear receptor Nur77 expression by n-butylenephthalide as pharmaceuticals on hepatocellular carcinoma cell therapy. Mol Pharmacol. 2008;74:1046–58.
Hossini AM, Quast AS, Plötz M, Grauel K, Exner T, Küchler J, et al. PI3K/AKT signaling pathway is essential for survival of induced pluripotent stem cells. PLoS One. 2016;11:e0154770.
Shi J, Li J, Guan H, Cai W, Bai X, Fang X, et al. Anti-fibrotic actions of interleukin-10 against hypertrophic scarring by activation of PI3K/AKT and STAT3 signaling pathways in scar-forming fibroblasts. PLoS One. 2014;9:e98228.
Lee TM, Lin SZ, Chang NC. Nicorandil regulates the macrophage skewing and ameliorates myofibroblasts by inhibition of RhoA/Rho-kinase signalling in infarcted rats. J Cell Mol Med. 2018;22:1056–69.
Liechty KW, Kim HB, Adzick NS, Crombleholme TM. Fetal wound repair results in scar formation in interleukin-10-deficient mice in a syngeneic murine model of scarless fetal wound repair. J Pediatr Surg. 2000;35:866–72.
Gordon A, Kozin ED, Keswani SG, Vaikunth SS, Katz AB, Zoltick PW, et al. Permissive environment in postnatal wounds induced by adenoviral-mediated overexpression of the anti-inflammatory cytokine interleukin-10 prevents scar formation. Wound Repair Regen. 2008;16:70–9.
Li L, Zhang S, Zhang Y, Yu B, Xu Y, Guan Z. Paracrine action mediates the antifibrotic effect of transplanted mesenchymal stem cells in a rat model of global heart failure. Mol Biol Rep. 2009;36:725–31.
Yang J, Dai C, Liu Y. A novel mechanism by which hepatocyte growth factor b locks tubular epithelial to mesenchymal transition. J Am Soc Nephrol. 2005;16:68–78.
Poli G. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Asp Med. 2000;21:49–98.
Wink DA, Vodovotz Y, Grisham MB, DeGraff W, Cook JC, Pacelli R, et al. Antioxidant effects of nitric oxide. Methods Enzymol. 1999;301:413–24.
Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. I. Paracrine cells important in health and disease. Am J Physiol. 1999;277:C1–9.
Jousset F, Maguy A, Rohr S, Kucera JP. Myofibroblasts electrotonically coupled to cardiomyocytes alter conduction: insights at the cellular level from a detailed in silico tissue structure model. Front Physiol. 2016;7:496.
Mias C, Coatrieux C, Denis C, Genet G, Seguelas MH, Laplace N, et al. Cardiac fibroblasts regulate sympathetic nerve sprouting and neurocardiac synapse stability. PLoS One. 2013;8:e79068.
Gyöngyösi M, Winkler J, Ramos I, Do QT, Firat H, McDonald K, et al. Myocardial fibrosis: biomedical research from bench to bedside. Eur J Heart Fail. 2017;19:177–91.
Lim GB. Heart failure: macrophages promote cardiac fibrosis and diastolic dysfunction. Nat Rev Cardiol. 2018;15:196–7.
Vallania F, Schiavone D, Dewilde S, Pupo E, Garbay S, Calogero R, et al. Genome-wide discovery of functional transcription factor binding sites by comparative genomics: the case of Stat3. Proc Natl Acad Sci USA. 2009;106:5117–22.
Sun Y, Iyer M, McEachin R, Zhao M, Wu YM, Cao X, et al. Genome-wide STAT3 binding analysis after histone deacetylase inhibition reveals novel target genes in dendritic cells. J Innate Immun. 2017;9:126–44.
Hutchins AP, Poulain S, Miranda-Saavedra D. Genome-wide analysis of STAT3 binding in vivo predicts effectors of the anti-inflammatory response in macrophages. Blood. 2012;119:e110–9.
Fu W, Hu W, Shi L, Mundra JJ, Xiao G, Dustin ML, et al. Foxo4- and Stat3-dependent IL-10 production by progranulin in regulatory T cells restrains inflammatory arthritis. FASEB J. 2017;31:1354–67.
Funding
This work was supported by the grants of An-Nan Hospital (ANHRF107-04) and Ministry of Science and Technology (MOST-106-2314-B-039-049), Taiwan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics approval
All rats had unrestricted access to food/water in accordance with the China Medical University Committee on Animal Care (Permit Number: 2016-070). The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Lee, TM., Harn, HJ., Chiou, TW. et al. Preconditioned adipose-derived stem cells ameliorate cardiac fibrosis by regulating macrophage polarization in infarcted rat hearts through the PI3K/STAT3 pathway. Lab Invest 99, 634–647 (2019). https://doi.org/10.1038/s41374-018-0181-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-018-0181-x
This article is cited by
-
Extracellular vesicles from adipose-derived stem cells promote microglia M2 polarization and neurological recovery in a mouse model of transient middle cerebral artery occlusion
Stem Cell Research & Therapy (2022)
-
Interplay between PI3K/AKT pathway and heart disorders
Molecular Biology Reports (2022)
-
Exosomes derived from human adipose mesenchymal stem cells attenuate hypertrophic scar fibrosis by miR-192-5p/IL-17RA/Smad axis
Stem Cell Research & Therapy (2021)
-
Intramyocardial injection of human adipose-derived stem cells ameliorates cognitive deficit by regulating oxidative stress–mediated hippocampal damage after myocardial infarction
Journal of Molecular Medicine (2021)
