
Abstract
Adipocyte-macrophage crosstalk plays a critical role to regulate adipose tissue microenvironment and cause chronic inflammation in the pathogenesis of obesity. Interleukin-29 (IL-29), a member of type 3 interferon family, plays a role in host defenses against microbes, however, little is known about its role in metabolic disorders. We explored the function of IL-29 in the pathogenesis of obesity-induced inflammation and insulin resistance. We found that serum IL-29 level was significantly higher in obese patients. IL-29 upregulated IL-1β, IL-8, and monocyte chemoattractant protein-1 (MCP-1) expression and decreased glucose uptake and insulin sensitivity in human Simpson-Golabi-Behmel syndrome (SGBS) adipocytes through reducing glucose transporter 4 (GLUT4) and AKT signals. In addition, IL-29 promoted monocyte/macrophage migration. Inhibition of IL-29 could reduce inflammatory cytokine production in macrophage-adipocyte coculture system, which mimic an obese microenvironment. In vivo, IL-29 reduced insulin sensitivity and increased the number of peritoneal macrophages in high-fat diet (HFD)-induced obese mice. IL-29 increased M1/M2 macrophage ratio and enhanced MCP-1 expression in adipose tissues of HFD mice. Therefore, we have identified a critical role of IL-29 in obesity-induced inflammation and insulin resistance, and we conclude that IL-29 may be a novel candidate target for treating obesity and insulin resistance in patients with metabolic disorders.
Similar content being viewed by others


Introduction
Obesity is considered as a serious global epidemic that significantly affects population health.1,2,3 According to the statistics of WHO in 2014, more than 600 million adults were obese in the world and it is further projected that 1.12 billion individuals will be classified as obese by 2030.4,5 The main cause is due to the imbalance between intake and consumption of calories, so that excessive energy stored in adipocytes leads to increase of cell numbers (hyperplasic adipocytes) and expansion of cell size (hypertrophic adipocytes). Obesity elevates the morbidity of cardiovascular disease, type 2 diabetes mellitus (T2DM), degenerative arthritis, and nonalcoholic fatty liver disease compared with healthy people.6,7
Hypertrophied adipocytes and adipose tissue-resident immune cells (primarily lymphocytes and macrophages) both contribute to higher proinflammatory cytokine production in the obese. The obesity-associated state of chronic low-grade systemic inflammation is considered a key step in the progression of insulin resistance and T2DM in humans and murine animal models.8,9,10,11 Previous study reported that tumor necrosis factor-α (TNF-α) levels were increased in obese adipose tissue and directly induced insulin resistance.12 Several studies have identified that the production of IL-1β, IL-6, and monocyte chemoattractant protein-1 (MCP-1) were elevated in obesity, which impaired insulin signaling indicating that inflammation is critical in the pathogenesis of obesity-induced metabolic disorders.13,14,15,16,17
Obesity is a low-grade chronic inflammation in adipose tissue. In the lean state, Th2 cells, Treg cells, eosinophils, and M2-like (anti-inflammatory) macrophages predominate in adipose tissue. Treg cells secrete IL-10 to maintain M2-like macrophages and inhibit macrophage migration. IL-4 released from Th2 cells and eosinophils and then induces the expression of IL-10 in M2-like macrophages, which in turn keep anti-inflammatory and insulin-sensitive phenotype. Hypertrophic adipocytes secrete MCP-1 to trigger the infiltration of monocytes and macrophages into the adipose tissue and then polarized to the M1-like (proinflammatory) macrophages.18 These immune cells release cytokines such as TNF-α, IL-1β, and IL-6 and then contribute to adipose tissue inflammation and insulin resistance.
Adipose tissue is recognized as a key organ in response to insulin. Binding of insulin to its receptor triggers the activation of insulin receptor substrate 1 (IRS1), which induces the downstream signaling cascades, including the phosphorylation of protein kinase B (also known as AKT), which regulates glucose transporter 4 (GLUT4) translocation into the plasma membrane.19 Several studies13,16,17,20 demonstrated that obesity is associated with an increased risk of developing insulin resistance and T2DM. Proinflammatory cytokines including TNF-α, IL-1β, and IL-6 affected the rate of glucose uptake and inflammation contributed to insulin resistance.
In 2002, two research groups identified newly three cytokines, IL-28A, IL-28B, and IL-29 (one of the group named IFN-λ2, IFN-λ3, and IFN-λ1).21,22 These cytokines belong to antiviral family of cytokines that are related to type I IFNs and IL-10 family members. IL-29-mediated signaling through a receptor complex contains IL-28R1 and IL-10R2 and then causes the activation of two tyrosine kinases, Janus kinase 1 and tyrosine kinase 2, which leads to the phosphorylation and activation of STAT1 and STAT2. IL-29 predominantly expressed in the epithelial tissues; however, several immune cells such as dendritic cells, macrophages, and Th17 cells are cellular sources of IL-29.23,24,25
IL-29 plays a critical role in host defense against microbes and is upregulated in viral-infected cells. IL-29 has an antitumor effect in several cancers including lung cancer, esophageal carcinomas, and colorectal cancer.26 In addition to its antiviral and antitumor activities, IL-29 acts as an immune modulator in rheumatoid arthritis and allergic asthma.27,28 However, little is known about the function of IL-29 in metabolic disorders. In the present study, we investigated whether IL-29 is involved in obesity-induced adipose tissue inflammation and insulin resistance.
Results
IL-29 is expressed in human adipose tissue and higher serum IL-29 levels were detected in obese patients
We examined whether IL-29 was involved in the pathogenesis of obesity and analyzed the IL-29 serum levels in obese patients and compared them with those of healthy individuals. According to body mass index (BMI) scale, 10 healthy individuals (BMI: 18.5-24.9 kg/m2), 13 pre-obese patients (overweight; BMI: 25.0-29.9 kg/m2), and 41 obese patients (BMI ≥ 30.0 kg/m2) were included in the analysis (Supplementary Table 1). Among obese patients, they were divided into obesity class I obesity (BMI: 30.0-34.9 kg/m2), class II obesity (BMI: 35.0-39.9 kg/m2), and class III obesity (BMI ≥ 40.0 kg/m2). We found that serum level of IL-29 was significantly higher in the patients with overweight and obesity than healthy individuals (Fig. 1a, b). The mean levels of serum IL-29 were 121.1 pg/ml in the healthy controls, 1100.9 pg/ml in the overweight, 1268.5 pg/ml in the obese class I, 1191.9 pg/ml in the obese class II patients, and 992.3 pg/ml in the obese class III patients. To clarify the clinical correlation between IL-29 and obesity, we also collected obese adipose tissue (visceral fat) and analyzed the expression of IL-29 and its receptors. Reverse-transcription polymerase chain reaction (RT-PCR) showed that IL-29 and its receptors were all expressed in adipose tissue isolated from obese patients (Fig. 1c). To further determine the cellular source of IL-29 in human visceral adipose tissue, we labeled tissue section with antibodies specific to IL-29 (Supplementary Fig. 1), fatty-acid binding protein-4 (FABP-4; adipocyte maker), and CD68 (macrophage marker). Immunofluorescence staining revealed that IL-29 was expressed in adipocytes and macrophages of obese patients (Fig. 1d, e and Supplementary Fig. 2). Fluorescence-activated cell sorting (FACS) analysis also showed that IL-29 and its receptors were expressed in visceral adipocytes isolated from obese patients (Fig. 1f).

Higher serum IL-29 levels in obese patients. a, b Level of IL-29 in serum from 10 healthy individuals, 13 overweight patients (pre-obese), 15 class I obese patients, 15 class II obese patients, and 11 class III obese patients were analyzed using ELISA. Data are expressed as mean ± SD of triplicate samples from a single experiment and are representative of three independent experiments. *P < 0.05 compared with healthy individuals. c The expression of IL-29 and its receptors (IL-28R1 and IL-10R2) in visceral adipose tissues isolated from obese patients was detected and analyzed using RT-PCR with specific primers. β-actin was an internal control. d Immunofluorescence staining for the adipocyte markers FABP-4 (red), IL-29 (green), and DAPI (blue) in the visceral adipose tissue of obese patients. Colocalization of IL-29 with FABP-4 is shown in yellow in the merged image. Scale bars = 30 μm. e Immunofluorescence staining for the macrophage markers CD68 (red), IL-29 (green), and DAPI (blue) in the visceral adipose tissue of obese patients. Colocalization of IL-29 with CD68 is shown in yellow in the merged image. Scale bars = 30 μm. f The expression of IL-29 and its receptors in visceral adipocytes isolated from obese patients was detected using FACS. Data are representative of three independent experiments
Upregulation of IL-29 receptors during SGBS adipocyte differentiation
To explore the role of IL-29 in adipocytes, we used a human cell line SGBS preadipocytes, which was originally obtained from an adipose tissue specimen of a patient with Simpson-Golabi-Behmel syndrome.29 It took 14 days to differentiate into mature SGBS adipocytes (Fig. 2a). We examined whether SGBS adipocytes are target cells for IL-29 signaling. Immunocytochemistry (IHC) staining and RT-qPCR showed that both IL-28R1 and IL-10R2 were expressed in SGBS adipocytes (Fig. 2b–d), which confirm our hypothesis. The expression of IL-28R1 and IL-10R2 were significantly increased during adipocyte differentiation (Fig. 2c, d). To explore the effect of IL-29 on adipogenesis, SGBS preadipocytes were incubated with IL-29 in the standard differentiation medium for 14 days. Although oil red O staining showed that IL-29 did not significantly affect adipogenesis (Fig. 2e), however, IL-29 downregulated adipogenic markers including peroxisome proliferator-activated receptor γ (PPARγ), FABP-4, and lipoprotein lipase (LPL) in mature SGBS cells in vitro (Fig. 2f, g). These data suggested that IL-29 is involved in the processes of lipid metabolism.

IL-29 reduced the expression of PPARγ, FABP-4, and LPL and increased IL-1β, IL-8, and MCP-1 expression in mature SGBS adipocytes. a The SGBS preadipocytes were differentiated into mature SGBS cells in differentiation medium for 14 days. The mature SGBS cells were stained with oil red O. b–d Expression of IL-28R1 and IL-10R2 in human mature SGBS adipocytes was analyzed using immunocytochemistry staining and RT-qPCR. *P < 0.05, **P < 0.01, ***P < 0.001 compared with day 0 controls. e Oil red O staining of mature SGBS adipocytes treated with or without IL-29 during differentiation for 14 days. f SGBS preadipocytes were differentiated into mature adipocytes in the absence or presence of IL-29 and the cell lysates of mature SGBS adipocytes were analyzed by immunoblotting with antibodies against PPARγ, FABP-4, and β-actin. β-actin was an internal control. g Human mature SGBS adipocytes were treated with IL-29 (100 ng/ml) for the indicated times. Total RNA was isolated for RT-qPCR with primers specific for LPL to amplify the transcripts. β-actin was an internal control. *P < 0.05 compared with 0 h controls. h–j Human mature SGBS adipocytes were treated with IL-29 (200-400 ng/ml) for 72 h. The conditioned medium was collected and the levels of IL-1β, MCP-1, and IL-8 were detected using ELISA. *P < 0.05, ***P < 0.001 compared with untreated controls. Data are expressed as mean ± SEM and are representative of three independent experiments. k Human mature SGBS adipocytes were treated with IL-29 (100 ng/ml) for 96 h in a hypoxic incubator. The conditioned medium was collected and the level of IL-8 was detected using ELISA. *P < 0.05 compared with untreated controls. Data are expressed as mean ± SEM and are representative of three independent experiments
IL-29 upregulated IL-1β, MCP-1, and IL-8 expression in mature SGBS adipocytes
To investigate the effect of IL-29 in adipocyte inflammation, mature SGBS adipocytes were treated with IL-29. RT-qPCR and enzyme-linked immunosorbent assay (ELISA) showed that the transcripts and protein levels of IL-1β, IL-8, and MCP-1 were increased in mature SGBS adipocytes after IL-29 treatment (Fig. 2h–j and Supplementary Fig. 3A–C). The similar results were found in primary human mature adipocytes isolated from obese patients (Supplementary Fig. 4A–F). To mimic the hypoxic conditions in adipose tissue with obesity, mature SGBS adipocytes were incubated with IL-29 in a hypoxic incubator for 96 h. ELISA showed that IL-29 increased IL-8 production in mature SGBS cells under hypoxic conditions (Fig. 2k).
IL-29 enhanced insulin resistance and impaired the insulin signaling
To clarify the role of IL-29 in glucose uptake in adipocytes, SGBS cells were treated with IL-29. RT-qPCR showed that IL-29 inhibited the expression of GLUT4 (Fig. 3a). Next, we further investigated the effect of IL-29 in insulin-stimulated glucose uptake. Mature SGBS adipocytes were treated with different concentration of IL-29 and then measured the uptake of 2-deoxyglucose in the basal state and after insulin stimulation. A glucose uptake assay indicated that IL-29 reduced insulin-stimulated glucose uptake in mature SGBS adipocytes (Fig. 3b). Furthermore, we treated mature SGBS adipocytes with IL-29 and then stimulated them with human insulin. Western blotting showed that IL-29 inhibited GLUT4 expression and suppressed AKT phosphorylation (Fig. 3c, d).

IL-29 promoted insulin resistance in mature SGBS adipocytes via reducing p-AKT and GLUT4 and enhanced monocyte chemotaxis. a Human mature SGBS adipocytes were treated with IL-29 (100 ng/ml) for the indicated times. Total RNA was isolated for RT-qPCR with primers specific for Glut4. **P < 0.01 compared with 0 h controls. β-actin was an internal control. b The differentiated SGBS mature adipocytes were incubated with IL-29 (10–200 ng/ml) in serum-free medium for 24 h, and then 2-deoxyglucose uptake was assessed using glucose uptake assay. **P < 0.01 compared with untreated controls. Data are expressed as mean ± SD and are representative of three independent experiments. c Human mature SGBS adipocytes were incubated with or without IL-29 (400 ng/ml) for 12 h and then stimulated with 0.2 μM insulin for 20 min. Cell lysates were collected and analyzed using immunoblotting with specific antibodies against phosphor-AKT and GLUT4. β-actin was an internal control. d The blot intensity was quantified using image j software. *P < 0.05, **P < 0.01 compared with untreated controls. e, f THP-1 monocytes were treated with IL-29 (100 ng/ml) for 4 h, and analyzed the migration activity using a modified Boyden chamber. Medium alone (0.5% FBS) was used as a negative control. FBS (5%) was used as a positive control. The number of migrated cells was counted. The results are expressed as a mean of four randomly selected fields. *P < 0.05 compared to untreated controls. Data are expressed as mean ± SEM and are representative of three independent experiments. g Normal chow diet (NCD) mice treated with or without IL-29 for 1 month. The total peritoneal cells from mice were isolated and stained with anti-F4/80 and then analyzed using FACS. Representative dot plot shows the peritoneal macrophages expressed F4/80. h Absolute numbers of F4/80+ macrophages in peritoneum of NCD mice. *P < 0.05 compared to PBS-treated controls. i Percentage of peritoneal macrophages F4/80+ cells. *P < 0.05 compared to PBS-treated controls. RT-qPCR analysis of MCP-1 expression in adipose tissues (j) and liver tissues (k) of NCD mice treated with or without IL-29 for 1 month. *P < 0.05 compared to PBS-treated controls. Data are expressed as mean ± SD and are representative of three independent experiments
IL-29 promoted monocyte migration
Monocytes migrate into adipose tissues is a critical process for the development of obesity. IL-29 was expressed in adipocytes and macrophages in adipose tissues isolated from obese patients. IL-29 promoted MCP-1 expression in adipocytes. Therefore, we hypothesize IL-29 might have a chemotactic property to recruit monocyte into adipose tissues. We performed migration assay to evaluate the effect of IL-29 on the migration of THP-1 monocytes using modified Boyden chamber. The result showed that IL-29 significantly promoted THP-1 monocyte migration in vitro (Fig. 3e, f). Previous studies21,22 reported that IL-29 was not present in the mouse genome; however, human IL-29 protein could function on mice through mouse IL-28R1 and IL-10R2 receptor complex. To further confirm the chemotactic property of IL-29, we treated human IL-29 into healthy BALB/c mice for 1 month and then isolated peritoneal exudate cells (PECs). FACS analysis showed that the numbers of F4/80+ macrophages was increased in IL-29-treated mice compared with the PBS-treated mice (Fig. 3g–i), which indicated that IL-29 promoted macrophage recruitment in vivo. In addition, RT-qPCR also showed that the expression of MCP-1 was upregulated in the liver and adipose tissues in IL-29-treated mice compared with the PBS-treated mice (Fig. 3j, k). Based on our finding that IL-29’s receptors (IL-28R1 and IL-10R2) were expressed in adipose tissue macrophages (ATM) and adipocytes of obese patients. These data raise the possibility that IL-29 plays a role in modulating macrophage and adipocyte in an autocrine and/or a paracrine manner.
Inhibition of IL-29 reduced IL-6, IL-8, and MCP-1 production in mature adipocyte-LPS-stimulated macrophage coculture system
Obesity is associated with an accumulation of macrophages in adipose tissues. This inflammation of adipose tissue is pivotal in the pathogenesis of several obesity-related disorders, particularly insulin resistance.6,30,31 A study reported that lipopolysaccharides (LPS) and IFN-α costimulated IL-29 expression in macrophages.32 We mimicked an macrophage-adipocyte in vitro model system of inflamed adipose tissue33 to elucidate the role of IL-29 in the microenvironment of adipose tissue with obesity (Fig. 4a). THP-1 monocytes were stimulated with PMA (100 ng/ml) to differentiate into macrophages, and then stimulated them with LPS and IFN-α to induce IL-29 production (Fig. 4b, c). Next, mature SGBS adipocytes were cocultured with these macrophages in the presence of anti-IL-29 monoclonal antibody (mAb) or control mIgG for 48 h. The conditioned medium was collected and analyzed. ELISA showed that anti-IL-29 mAb not only reduced IL-29 but also decreased the levels of IL-6, IL-8, and MCP-1 compared with the mIgG-treated controls (Fig. 4d). Anti-IL-29 mAb also downregulated the transcripts of IL-1β, IL-6, IL-8, and MCP-1 in mature SGBS cells compared with the mIgG-treated controls (Fig. 4e). These data suggested that IL-29 plays a regulatory role in crosstalk between macrophages and adipocytes.

Inhibition of IL-29 reduced IL-6, IL-8, and MCP-1 production in mature adipocyte-LPS-stimulated macrophage coculture system. a Schematic representation of in vitro inflamed adipose tissue model. b The expression of IL-29 and its receptors in monocytes, macrophages, and LPS-induced macrophages. c The level of IL-29 in coculture system was measured using ELISA. *P < 0.05 compared with SGBS only group. d Levels of IL-6, IL-8, MCP-1, and IL-29 in coculture system were detected using ELISA after anti-IL-29 mAb (1.5 μg/ml) or control mIgG (1.5 μg/ml) treatment for 48 h. *P < 0.05, **P < 0.01 compared with the mIgG-treated group. e After coculture for 48 h, the total RNA from SGBS cells was isolated for RT-qPCR with primers specific for IL-1β, IL-6, IL-8, and MCP-1 to amplify the transcripts. β-actin was an internal control. *P < 0.05, **P < 0.01 compared with the mIgG-treated group. Data are expressed as mean ± SEM and are representative of three independent experiments
IL-29 promoted obesity-induced insulin resistance in obese mice
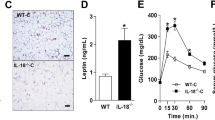
Next, we examined the effects of IL-29 on obesity-induced inflammation and insulin resistance in vivo. C57BL/6 male mice were fed normal chow diet (NCD) until they were 6 weeks old. Subsequently, they were randomly assigned to the NCD or high-fat diet (HFD) group for 16 weeks. The treatment began after the 1-week HFD, at which time the mice were divided into three groups: (i) HFD mice treated with PBS, (ii) HFD mice treated with 0.5 mg of IL-29/kg/twice a week, and (iii) HFD mice treated with 1 mg of IL-29/kg/twice a week. Changes in body weight were measured weekly. Body weights were not significantly different between groups at the beginning, but after 6 weeks, body weight was higher during week 6-10 in the IL-29-treated mice (0.5 mg/kg) than in the PBS-treated mice (Fig. 5a), despite equivalent total caloric intakes (data not shown). IL-29 slightly influences the body weight gain and fat pad weight in the HFD-fed mice after treatment for 16 weeks (Fig. 5b). However, we found that IL-28R1 was upregulated in white adipose tissues (Fig. 5c). Moreover, glucose and insulin tolerance tests (ITTs) showed that IL-29-treated HFD mice had worsened glucose homeostasis and insulin responsiveness than did PBS-treated HFD mice (Fig. 5d, e). Western blotting also showed that insulin-regulated glucose transport pathway GLUT4 and phosphor-AKT were downregulated in adipose tissues of IL-29-treated HFD mice compared with the PBS-treated HFD mice (Fig. 5f).

IL-29 enhanced obesity-induced insulin resistance and increased macrophage number in HFD mice. C57BL/6 mice were randomly assigned to the NCD (10% kcal derived from fat) or the high-fat diet (HFD; 60% kcal derived from fat) group for 16 weeks. HFD mice were divided into three groups (n = 5 mice per group): HFD mice without treatment, HFD mice treated with 0.5 mg/kg IL-29, and HFD mice treated with 1 mg/kg IL-29 twice a week. a Body weight was measured weekly. b The fat pad weight and weight gain were measured after 16 weeks. Data are expressed as mean ± SEM and are representative of three independent experiments. c RT-qPCR analysis of IL-28R1 and IL-10R2 mRNA expression in the visceral white adipose tissue (WAT) of HFD mice and NCD mice after 16 weeks. *P < 0.05 compared with NCD mice. d A glucose tolerance test (GTT) and e an insulin tolerance test (ITT) was performed at weeks 15 and 16, respectively, after IL-29 treatment. *P < 0.05 compared with PBS-treated HFD mice. f The adipose tissues from PBS-treated and IL-29-treated HFD mice were analyzed using immunoblotting with specific antibodies against phosphor-AKT and GLUT4. β-actin was an internal control. g Total peritoneal cells from PBS-treated and IL-29-treated HFD mice were stained with anti-CD11c, anti-F4/80, and anti-CD206 and analyzed by FACS. A dot plot defining the F4/80+ cells in the peritoneum of HFD mice. F4/80+ cells were quantified by flow cytometry. h, i The absolute cell numbers and total percentage of F4/80+ macrophages in peritoneum of PBS-treated and IL-29-treated HFD mice. *P < 0.05 compared with PBS-treated HFD mice. j, k Expression of CD11c and CD206 was measured as mean fluorescence intensities (MFIs) by flow cytometry. l–o F4/80, CD11c, CD206, and MCP-1 mRNA expression in adipose tissues isolated from PBS-treated and IL-29-treated HFD mice were analyzed using RT-qPCR with specific primers. *P < 0.05 compared with PBS-treated HFD mice. p Ratio of CD11c+ cells relative to resident CD206+ cells was calculated in MFI units. Data are expressed as mean ± SEM and are representative of three independent experiments. *P < 0.05 compared with PBS-treated HFD mice. q The numbers of CD11b+Ly6G+ neutrophils in adipose tissue of PBS-treated and IL-29-treated HFD mice. Data are expressed as mean ± SEM. *P < 0.05 compared with the PBS-treated HFD mice
IL-29 increased macrophage recruitment and adipose tissue inflammation in obese mice
We further analyzed the number of macrophages in the PECs of HFD mice. FACS analysis showed that the number and percentage of total macrophages (F4/80+) was increased in IL-29-treated HFD mice (Fig. 5g–i). The expression of CD11c on M1 macrophages (F4/80+CD206−) was increased (Fig. 5j), but there was no significant change of CD206 on M2 macrophages (F4/80+CD11c−) in IL-29-treated HFD mice compared with the PBS-treated HFD mice (Fig. 5k). RT-qPCR showed that the expression of F4/80, CD11c, and MCP-1 was upregulated, but CD206 was not changed in adipose tissue of IL-29-treated HFD mice (Fig. 5l–o). FACS analysis confirmed that CD11c+/CD206 ratio was higher in adipose tissue of IL-29-treated HFD mice compared with the PBS-treated HFD mice (Fig. 5p). In addition, we observed that the number of adipose tissue neutrophils was increased in IL-29-treated HFD mice compared with PBS-treated HFD mice (Fig. 5q). To further confirm the role of IL-29 in adipose tissue inflammation, mature adipocytes isolated from visceral adipose tissue of wild-type (WT) and IL-28R1 deficiency mice were incubated with and without IL-29. RT-qPCR showed that IL-29 increased the expression of IL-1β and MCP-1 in adipocytes from WT mice (Supplementary Fig. 5A, B). IL-28R1 deficiency abolished IL-29-induced IL-1β and MCP-1 expression in IL-29-treated adipocytes (Supplementary Fig. 5C, D), which suggested that IL-28R1-mediated IL-29 signaling is critical for IL-29-induced adipose tissue inflammation.
Discussion
Adipose tissue inflammation is a key process in the development of obesity-induced insulin resistance. In this study, we found that serum IL-29 was significantly higher in obese patients. IL-29 was expressed in ATM and adipocytes in obese patients. IL-29 promoted adipose tissue inflammation, macrophage chemotaxis, and systemic insulin resistance. Moreover, IL-29 treatment in vivo promoted inflammation, insulin resistance, and macrophage recruitment in adipose tissues. Collectively, these findings suggest that IL-29 secretion by adipocytes that promote ATMs retention in visceral WAT and foster the chronic inflammation that leads to metabolic disorder.
In the present study, we found that IL-29 receptors (IL-28R1 and IL-10R2) were significantly increased during adipocyte differentiation, which allowed us to explore the role of IL-29 in adipogenesis. Our results showed that IL-29 did not significantly affect adipogenesis; however, IL-29 inhibited the expression of PPARγ, FABP-4, and LPL in mature SGBS cells. Previous studies34,35,36,37 reported that PPARγ is a master regulator of adipogenesis and directly controls many genes that are critical for maintaining the functions of adipocytes including lipid transport (FABP-4), fatty-acid uptake (LPL), and insulin signaling (GLUT4). Therefore, our findings suggested that IL-29 is involved in the processes of lipid metabolism. Inflammatory factors play a critical role in the development of dyslipidemia and they also stimulate lipolysis in adipocytes.38,39 TNF-α, IL-1β, and IL-6 enhanced lipolysis and suppressed activity of LPL, which is a key regulatory enzyme in the catabolism and clearance of triglyceride-rich lipoproteins in adipocytes.40,41,42,43,44 In our study, we found that IL-29 decreased LPL expression in adipocytes. Further studies are required to investigate whether IL-29 involved in the development of dyslipidemia to support the connection between IL-29 and obesity-related metabolic diseases.
Various cytokines such as TNF-α, IL-1β, IL-6, and IL-8 are involved in the development of obesity and contributed both to local and systemic inflammation, thus potentiating insulin resistance.9,11 IL-29 increased IL-1β, MCP-1, and IL-8 levels in mature SGBS adipocytes and human primary mature adipocytes. In addition, IL-29 increased IL-8 protein level in mature SGBS adipocytes under hypoxic conditions. IL-8 is known as neutrophil chemotactic factor and induces chemotaxis in other cell types including macrophages.14,45,46,47 Our results suggested that IL-29 might play a role for promoting inflammatory responses and enhancing neutrophil chemotaxis to adipose tissue through regulating IL-8 under hypoxic conditions. In addition, IL-29 impaired insulin-mediated glucose uptake through inhibition of insulin-stimulated GLUT4 expression and impaired insulin signaling through reducing phosphorylation of AKT. Therefore, IL-29 might directly affect local inflammation and insulin resistance, and indirectly affect these activities by inducing other mediators via autocrine/paracrine signaling.
Obesity is associated with an increased numbers of macrophages in adipose tissue. We used an in vitro macrophage-adipocyte coculture model to mimic a microenvironment in obese adipose tissues and found that blockade of IL-29 activity reduced the production of proinflammatory cytokines IL-6, IL-8, and MCP-1. Therefore, these findings suggested that IL-29 and other proinflammatory cytokines synergistically mediate the initial inflammatory response in adipose tissues. Furthermore, MCP-1 binding to its receptor, CCR2, is important in a variety of infectious and inflammatory diseases.48 The MCP-1/CCR2 system contributes to monocyte migration into adipose tissues. IL-29 promoted MCP-1 expression in the liver and adipose tissues of obese mice. It is possible that IL-29 might indirectly enhance the migration of monocytes through the MCP-1-CCR2 axis in obesity. Previous studies49,50 reported that IL-28A had an anti-inflammatory effect by restricting recruitment of IL-1β-expressing neutrophils in arthritis mouse model. In addition, IL-29 exerted an anti-inflammatory function in vitro by inhibiting the formation of neutrophil extracellular traps and the cytoplasmic expression of tissue factor in neutrophils. In the current study, our results showed that IL-29 promoted adipose tissue inflammation and increased the number of neutrophils in adipose tissue of HFD mice. The discrepancy of IL-29 function might be due to the different animal models. Whether IL-29 directly regulates neutrophil function in adipose tissues requires additional investigation.
No significant difference in body weight was observed in IL-29-treated HFD mice for the initial 5 weeks. However, body weight was higher in IL-29-treated HFD mice than in PBS-treated HFD mice during weeks 6-10. We hypothesized that other cytokines compensated for the activity of IL-29 during the early acute inflammatory response. IL-29 might be more critical for monocyte/macrophage recruitment and regulating glucose homeostasis in the chronic inflammatory state and contribute to metabolic dysfunction.
IL-10 is an anti-inflammatory cytokine in adipose tissue.51 IL-10 signals through a receptor complex consisting of IL-10R1 and IL-10R2.52 IL-29 is also a member of IL-10 family because its amino acid sequence and structure are conserved to IL-10.21,22 IL-29 signals through IL-28R1 and IL-10R2, and acts as a proinflammatory cytokine in adipose tissue. IL-10R2 is a common receptor for IL-10 and IL-29. The regulation between IL-10 and IL-29 in metabolic homeostasis and in adipose tissues is not clear. Previous study53 reported that IL-29 enhanced sensitivity of macrophage to IL-10 through upregulating IL-10R1 expression; however, blocking of IL-10R1 signal did not influence the regulation of TLR-induced IL-12 production in IL-29-primed macrophages. These data indicated that IL-10 and IL-29 induced distinct signaling pathways in macrophages. Furthermore, IL-29 enhanced IFNγ-induced IL-12 and TNF production by monocyte-derived macrophages in response to TLR stimulation through upregulating IFNγR1 expression.54 IL-12 played a critical role in the differentiation of Th1 cells and also promoted macrophage migration and differentiated into the proinflammatory M1-like macrophages.55 In contrast to macrophage, IL-29 also promoted Th1 cytokine IFN-γ expression and suppressed Th2 polarization.56 Taken together, these studies provided more evidence that IL-29 might create a favorable microenvironment for macrophage-adipocyte crosstalk to promote adipose tissue inflammation in obesity.
Neutralization of the activity of cytokines is a strategy for reducing obesity-induced inflammation and insulin resistance. Some studies57,58 found that anti-TNF-α antibodies reduced blood glucose in obese individuals but were ineffective for improving insulin sensitivity. Anti-IL-1β antibodies improved glucose homeostasis, but not insulin sensitivity.59,60 We found that IL-29 inhibited insulin-stimulate glucose uptake in adipocytes, which indicated that inhibition of IL-29 might not only reduce inflammation response, but also improve insulin sensitivity. IL-29 may contribute to the metabolic complications associated with obesity such as T2DM and dyslipidemia by altering adipose tissue function and promoting adipose tissue inflammation.
In summary, our findings demonstrate that IL-29 is involved in metabolic homeostasis. IL-29 is an important regulator in adipocyte-macrophage crosstalk and promoted macrophage recruitment and insulin resistance. Therefore, we have identified a critical role of IL-29 in obesity-induced inflammation and insulin resistance. We conclude that IL-29 is a novel candidate target for treating obesity-induced inflammation and insulin resistance.
Materials and methods
Patients and samples
World Health Organization criteria were used to categorize the participants into several groups based on the participant’s BMI. We collected blood samples from 41 obese patients, 13 pre-obese patients, and 10 healthy individuals from 2013 to 2016 in National Cheng Kung University Hospital, Tainan, Taiwan. Blood was centrifuged (2000 rpm for 10 min at 4 °C), and serum was collected for ELISA analysis. Levels of IL-29 in the serum of the participants were determined using human ELISA kit (BioLegend, San Diego, CA, USA) according to the manufacturer’s instructions. Adipose tissues were harvested from three obese patients. RNA extract from obese adipose tissue were used for RT-PCR analysis. Signed informed consent was obtained from all participants. Written informed consent was obtained. The National Cheng Kung University Hospital Institutional Review Board approved the study (IRB number: A-ER-102-052). The study was done in accordance with approved guidelines.
Generation of anti-IL-29 mAb
Anti-IL-29 mAb (164B) was generated following standard protocols. Full length of human IL-29 protein was used as an antigen to immunize mice. Briefly, BALB/c mice were subcutaneously immunized with human IL-29 recombinant protein (50 μg/mouse) emulsified with an equal volume of Freund’s complete/incomplete adjuvant, and boosted by an intravenous injection of the antigen without adjuvant 3 days before fusion. Spleen cells from immunized mice were fused with Sp2/0-Ag14 myeloma cells with poly ethylene glycol (PEG 1500; Sigma-Aldrich, St. Louis, MO). The selection and characterization of anti-IL-29 mAbs were based on the standard protocols. The isotype was determined using an isotyping ELISA assay (R&D Systems, Minneapolis, MN, USA). The isotype of anti-IL-29 mAb (clone 164B) was IgG2a. The mAbs were purified from ascites by using protein-A column and fast liquid chromatography (FPLC, GE Healthcare, Illinois, USA).
Isolation of primary mature adipocytes
Visceral adipose tissues from obese patients were digested by 1 mg/ml type II collagenase (Sigma-Aldrich) for 30-45 min at 37 °C and then centrifuged for 1 min at 500 rpm. After the centrifugation, the mature adipocytes floated in the top layer of the solution. The top layer of the solution was transferred into a new tube and washed with DMEM/F12 containing 1% heat-inactivated fetal bovine serum (FBS; Hyclone, GE Healthcare Life Sciences) twice. The isolated mature adipocytes were diluted in 1:10 ratio for further experiments. Mouse primary mature adipocytes were isolated following the same protocols.
Immunofluorescence staining
The localization of IL-29 was assessed using immunofluorescence staining of endogenous IL-29 and co-staining with a specific marker for adipocytes and macrophages. Paraffin-embedded human adipose tissue samples were deparaffinized and blocked in antibody diluent (Dako#S3022, Carpinteria, CA, USA) for 1 h at room temperature and then incubated with anti-CD68 (1:100; BD Biosciences Pharmingen, San Diego, CA), anti-FABP-4 (1:200; Proteintech group, Chicago, IL, USA), and anti-IL-29 (5 μg/ml; clone 164B) antibodies at 4 °C overnight. Isotype-matched control antibodies were used as negative control. The samples were washed with PBS and stained with the corresponding Alexa-Fluor-coupled secondary antibodies (Invitrogen) at a 1:500 dilution to detect the bound antibodies. The slides were mounted with ProLong® Gold antifade reagent with DAPI (Invitrogen). Images were taken using scanning confocal laser microscopy (Olympus FV1000) to visualize the stained cells.
SGBS cell differentiation and treatment
SGBS preadipocytes were kindly provided by Professor Martin Wabitsch (University of Ulm, Germany). Human SGBS preadipocytes were cultured in DMEM/F12 medium containing 10% fetal calf serum (FCS; Invitrogen), 3.3 μM biotin (Sigma-Aldrich), 1.7 μM pantothenate (Sigma-Aldrich), 100 units/ml of penicillin (Caisson), and 100 µg/ml streptomycin (Caisson) until reaching confluence. To induce SGBS preadipocytes differentiation, cells were washed with PBS twice and cultured in differentiation medium (DMEM/F12 supplemented with 3.3 μM biotin, 1.7 μM pantothenate, 2 μM rosiglitazone, 25 nM dexamethasone, 0.5 mM methylisobuthylxantine (IBMX), 1 µM cortisol, 0.01 mg/ml transferrin, 0.2 nM triiodotyronin, and 20 nM human insulin) for the first 4 days. After 4 days, the differentiating cells were kept in differentiation medium excluding dexamethasone, IBMX, and rosiglitazone. The medium containing 400-800 ng/ml of IL-29 (R&D Systems) was changed every 4 days.
Oil red O staining
SGBS preadipocytes were incubated for 14 days in differentiation medium. Mature SGBS adipocytes were washed twice with PBS and fixed with 3.7% formaldehyde for 30 min. After fixation, the cells were washed twice with distilled water and once with 1, 2-isopropanediol (Sigma). Then, the cells were stained with oil red O (Sigma) solution (0.3 g oil red O in 100 ml 60% 1,2-isopropanediol) for 15-30 min at room temperature. Cells were visualized through 4× or 10× objectives mounted on for observation. For the quantification, the oil red O dye was extracted with 100% isopropanol for 5 min. Equal amounts of elution from each well were transferred to a 96-well plate, and, using a microplate reader (Multiskan Spectrum; Thermo Scientific, Vantaa, Finland), the absorbance values were measured at a wavelength of 492nm.
IHC staining
Cells were rinsed with PBS twice and fixed in 3.7% formaldehyde for 30 min. Cells were then blocked in antibody diluent (#S3022, Dako, Carpinteria, CA, USA) for 2 h at room temperature and incubated with anti-IL-28R1 (1:200; Bioss Antibodies Inc., Woburn, MA, USA) and anti-IL-10R2 (1:200; Bioss Antibodies Inc.) polyclonal antibodies at 4 °C overnight. Incubating paraffin tissue sections with rabbit IgG (ab37415, Abcam, Cambridge, MA, USA) instead of primary antibody was the negative control. The samples were washed with PBS and incubated with appropriate secondary antibody (Invitrogen). The immune reactivity of positive staining was developed by using the 3-amino-9-ethylcarbazole (AEC) chromogen kit (Romulin AEC Chromogen Kit; Biocare Medical, Walnut Creek, CA) and counterstained with Mayer’s hematoxylin (J. T. Baker, Phillipsburg, NJ).
Reverse-transcription polymerase chain reaction
The total RNA of cells and adipose tissue samples was isolated (Invitrogen, Carlsbad, CA) and reverse transcription was done (PrimeScript RT-PCR kit; Clontech, Palo Alto, CA). The expression of IL-29, IL-28R1, and IL-10R2 was analyzed using PCR with gene-specific primers (Supplementary Table 2). β-actin was used as an internal control.
Quantification real-time PCR (Real-time qPCR/RT-qPCR)
Total RNA was isolated and reverse transcription was done. RT-qPCR was performed with a Rotor-Gene Q detection system (QIAGEN) with gene-specific primers (Supplementary Table 2). Quantification analysis of messenger RNA was normalized with β-actin, which was used as the housekeeping gene. Relative multiples of changes in mRNA expression were determined by calculating 2–ΔΔCt.
Enzyme-linked immunosorbent assay
To test the specificity of anti-IL-29 mAb (164B), cytokines of the IL-10 family (IL-10, -19, -20, -22, -24, -28, and -29) were coated on the plate with 1 μg/ml and analyzed for their binding with 0.4 μg/ml anti-IL-29 mAb (164B) using direct ELISA. The levels of IL-6, IL-8, and MCP-1 in IL-29-treated mature SGBS adipocytes, IL-29-treated human primary mature adipocytes, and in macrophage-adipocyte coculture conditioned medium were measured using ELISA kits (R&D Systems) according to the manufacturer’s instructions.
Western blotting
Proteins were separated by SDS-PAGE and transferred electrophoretically to PVDF membranes (Millipore, USA). Membranes were blocked with 5% (w/v) nonfat milk in PBST for 2 h at room temperature, and then incubated overnight at 4 °C with primary antibodies: anti-FABP-4 (1:1000; Proteintech group), anti-PPARγ (1:1000; Proteintech Group), anti-GLUT4 (1:1000; Cell Signaling), anti-phosphoAkt (1:1000; Cell Signaling), and anti-β-actin (1:10000; Proteintech Group). After binding of primary antibodies, membranes were washed with PBST three times and incubated for 1 h at room temperature with the species-specific horseradish peroxidase-labeled secondary antibodies. Binding of secondary antibodies was detected with SuperSignal West Pico Chemiluminescent Substrate where the chemiluminescent signals were visualized and imaged following exposure and development of Hyperfilm ECL molecular on luminescence imaging system.
Hypoxia treatment
Hypoxic culture conditions were established using an O2/CO2 incubator (ASTEC, Fukuoka, Japan). Mature SGBS adipocytes were incubated in a low-oxygen condition consisting of 1% oxygen, 5% carbon dioxide, and 94% nitrogen for 96 h. The conditioned medium was collected and analyzed using ELISA.
Glucose uptake assay
SGBS preadipocytes were cultured in 96-well plates and differentiated into mature adipocytes. Mature SGBS adipocytes were treated with IL-29 (10-200 ng/ml) (R&D Systems) for 12 h and then measured the rate of glucose uptake. The glucose uptake assays were performed using the Glucose Uptake Colorimetric Assay Kit (BioVision, USA) according to the manufacturer’s instructions.
Cell migration assay
THP-1 cells were assayed using a modified Boyden chamber housing a polycarbonate filter with 8-mm pores (Nucleopore, Cabin John, MD). The upper wells were loaded with 2.5 × 104 THP-1 cells. The lower chambers were filled with IL-29 (100 ng/ml) in RPMI 1640 medium containing 0.5% FBS. RPMI 1640 medium with 0.5% FBS was used as a negative control; 5% FBS was used as a positive control. The chambers were incubated for 4 h at 37 °C. Cells adhering to the lower side of the filter were fixed with methanol and stained in a Giemsa solution (Diff-Quick; Baxter Healthcare, Deerfield, IL) for counting. The experiment was carried out three times using quadruplicate wells. Migrated cells were counted in four randomly selected fields of 100-fold magnification.
Coculture of mature SGBS adipocytes with macrophages
THP-1 monocytes (American Type Culture Collection, USA) were cultured in RPMI-1640 medium with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate (Caisson), 2.5 g/L D-glucose, and 0.05 nM β-mercaptoethanol (Invitrogen, USA). In the coculture experiments, THP-1 monocytes were seeded in six transwell inserts (SPL, membrane pore size of 0.4 μm) and differentiated into macrophages with 100 nM phorbol 12-myristate 13-acetate (PMA) for 48 h. Monocyte-derived macrophages were pretreated with 100 IU IFN-α (PeproTech) for 16 h, and then added LPS (1 μg/ml) to stimulate the expression of IL-29. After stimulation, inserts were transferred to the six-well plates containing mature SGBS adipocytes and incubated with 1.5 μg/ml of anti-IL-29 mAb (R&D Systems, #247801) or control mIgG (R&D Systems, #20102) for another 48 h. The cultured conditioned medium was collected for ELISA analysis and the mRNA were isolated from SGBS cells for RT-qPCR analysis.
Animals
All animal experiments were conducted according to the protocols based on the Taiwan National Institutes of Health (Taipei, Taiwan) standards and guidelines for the care and use of experimental animals. The research procedures were approved by the Animal Ethics Committee of National Cheng Kung University (IACUC approval no. 106241). The methods were carried out in accordance with the approved guidelines. All efforts were made to minimize animal suffering and to reduce the number of animals used. IL-28R1 deficiency (IL-28R1−/−) mice were kindly supplied by Professor Peter Staeheli (Institute of Virology, Medical Center University of Freiburg, Germany). BALB/c and C57BL/6 mice were purchased from the National Laboratory Animal Center (Taipei, Taiwan) and kept on a 12-h light-dark cycle at 22 ± 2 °C. Body weight of the animals was recorded weekly. Eight-week old C57BL/6 mice were given NCD or HFD (60% of calories, D12492, Research Diets Inc., NJ, USA) for 16 weeks. HFD mice were randomly divided into three groups (n = 5 mice/group): HFD mice without treatment, HFD mice treated with 0.5 mg/kg, or 1 mg/kg of IL-29 twice a week. Body weight changes were measured weekly.
Glucose tolerance test (GTT) and ITT
GTT and ITT tests were performed after 14 and 15 weeks of treatment, respectively. For GTT, the mice were fasted overnight and intraperitoneally (i.p.) injected with 2 g/kg D-glucose. Blood samples were collected from the tail tip. Blood glucose levels were measured at 0 (before injection), 30, 60, 90, and 120 min after injection using an Accu-chek glucose meter (Roche Diagnostics). For the insulin tolerance test, the mice fasted for 12 h, and were then i.p. injected with recombinant insulin (0.5 unit/kg). Blood glucose was similarly measured.
FACS analysis
Mature adipocytes isolated from obese patients were incubated with Human BD Fc Block (BD Biosciences) for 10 min and then stained with primary antibodies including IL-28R1 (0.5 μg/ml; Abcam) and IL-10R2 (0.5 μg/ml; R&D Systems). Then the cells were washed twice and incubated with Alexa 488-conjugated goat anti-rabbit IgG secondary antibody (Invitrogen) at 4 °C for 30 min. Cells were washed and immersed in 0.5 ml of buffered formalin (BD Biosciences) at room temperature for 30 min. One milliliter of permeabilization buffer (BD Biosciences) was added and the cells were collected by centrifugation. The cells were washed with permeabilization buffer and resuspended in 100 μl of permeabilization buffer containing mouse anti-human IL-29 mAb (0.5 μg/ml; 164B) at 4 °C for 30 min. Then the cells were washed twice with permeabilization buffer and then incubated with Alexa 647-conjugated goat anti-mouse IgG2a secondary antibody (Invitrogen) at 4 °C for 30 min. The cells were resuspended in FACS buffer (PBS containing 5% FBS and 2 mM EDTA) and analyzed using a FACSCalibur (BD Biosciences). Human mature adipocytes were identified by forward scatter and side scatter as previously described.61 For mouse model, adipose tissues from mice were digested with 1 mg/ml type II collagenase (Sigma-Aldrich) for 30 min at 37 °C and passed through a 70-μm cell strainers (BD Biosciences). Red blood cells were lysed using ACK buffer and centrifuged at 1500 rpm for 10 min. Cells were washed twice with FACS buffer and counted using a Nexcelom cell counter. Cells were incubated with Mouse BD Fc Block (BD Biosciences) for 10 min and then stained with 0.2 μg/ml of APC-conjugated anti-F4/80, PE-conjugated anti-CD11c, and FITC-conjugated anti-CD206 (BD Biosciences) for analyzing macrophages and also stained with 0.2 μg/ml of PerCP-conjugated anti-CD11b and FITC-conjugated anti-Ly6G (BD Biosciences) for analyzing neutrophils. The data were collected and analyzed using a FACSCalibur.
Statistical analysis
Prism 7.0 (GraphPad Software) was used for the statistical analysis. T-test and a one-way ANOVA nonparametric test (Kruskal–Wallis test) were used to compare the data between groups. Data are expressed as the mean of replicate measurements or mean normalized values between multiple experiments ± SEM or SD. P < 0.05 was considered statistically significantly.
References
Nguyen, D. M. & El-Serag, H. B. The epidemiology of obesity. Gastroenterol. Clin. N. Am. 39, 1–7 (2010).
Collaboration NCDRF. Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 387, 1377–1396 (2016).
Maffetone, P. B., Rivera-Dominguez, I. & Laursen, P. B. Overfat and underfat: new terms and definitions long overdue. Front. Public Health 4, 279 (2016).
Kelly, T., Yang, W., Chen, C. S., Reynolds, K. & He, J. Global burden of obesity in 2005 and projections to 2030. Int J. Obes. (Lond.) 32, 1431–1437 (2008).
Patel, D. Pharmacotherapy for the management of obesity. Metabolism 64, 1376–1385 (2015).
Guilherme, A., Virbasius, J. V., Puri, V. & Czech, M. P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 9, 367–377 (2008).
Kim, J. B. Dynamic cross talk between metabolic organs in obesity and metabolic diseases. Exp. Mol. Med. 48, e214 (2016).
Shoelson, S. E., Lee, J. & Goldfine, A. B. Inflammation and insulin resistance. J. Clin. Investig. 116, 1793–1801 (2006).
Gregor, M. F. & Hotamisligil, G. S. Inflammatory mechanisms in obesity. Annu Rev. Immunol. 29, 415–445 (2011).
Ouchi, N., Parker, J. L., Lugus, J. J. & Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97 (2011).
Hotamisligil, G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 542, 177–185 (2017).
Hotamisligil, G., Shargill, N. & Spiegelman, B. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91 (1993).
Kern, P. A., Ranganathan, S., Li, C., Wood, L. & Ranganathan, G. Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 280, E745–E751 (2001).
Kim, C. S. et al. Circulating levels of MCP-1 and IL-8 are elevated in human obese subjects and associated with obesity-related parameters. Int J. Obes. (Lond.) 30, 1347–1355 (2006).
Moschen, A. R. et al. Adipose and liver expression of interleukin (IL)-1 family members in morbid obesity and effects of weight loss. Mol. Med. 17, 840–845 (2011).
Vandanmagsar, B. et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 (2011).
Panee, J. Monocyte chemoattractant protein 1 (MCP-1) in obesity and diabetes. Cytokine 60, 1–12 (2012).
Lackey, D. E. & Olefsky, J. M. Regulation of metabolism by the innate immune system. Nat. Rev. Endocrinol. 12, 15–28 (2016).
Khan, A. & Pessin, J. Insulin regulation of glucose uptake: a complex interplay of intracellular signalling pathways. Diabetologia 45, 1475–1483 (2002).
Rotter, V., Nagaev, I. & Smith, U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J. Biol. Chem. 278, 45777–45784 (2003).
Sheppard, P. et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 4, 63–68 (2003).
Kotenko, S. V. et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 4, 69–77 (2003).
Wolk, K. et al. Maturing dendritic cells are an important source of IL-29 and IL-20 that may cooperatively increase the innate immunity of keratinocytes. J. Leukoc. Biol. 83, 1181–1193 (2008).
Wolk, K. et al. IL-29 is produced by T(H)17 cells and mediates the cutaneous antiviral competence in psoriasis. Sci. Transl. Med. 5, 204ra129 (2013).
Siren, J., Pirhonen, J., Julkunen, I. & Matikainen, S. IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J. Immunol. 174, 1932–1937 (2005).
Kelm, N. E. et al. The role of IL-29 in immunity and cancer. Crit. Rev. Oncol. Hematol. 106, 91–98 (2016).
Wang, F. et al. Interleukin-29 modulates proinflammatory cytokine production in synovial inflammation of rheumatoid arthritis. Arthritis Res. Ther. 14, R228 (2012).
Li, Y. et al. Adenovirus expressing IFN-lambda1 (IL-29) attenuates allergic airway inflammation and airway hyperreactivity in experimental asthma. Int. Immunopharmacol. 21, 156–162 (2014).
Wabitsch, M. et al. Characterization of a human preadipocyte cell strain with high capacity for adipose differentiation. Int J. Obes. Relat. Metab. Disord. 25, 8–15 (2001).
Weisberg, S. P. et al. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 112, 1796–1808 (2003).
Lumeng, C. N., Bodzin, J. L. & Saltiel, A. R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 117, 175–184 (2007).
Sirén, J., Pirhonen, J., Julkunen, I. & Matikainen, S. IFN-α Regulates TLR-Dependent Gene Expression of IFN-α, IFN-β, IL-28, and IL-29. J. Immunol. 174, 1932–1937 (2005).
Keuper, M., Dzyakanchuk, A., Amrein, K. E., Wabitsch, M. & Fischer-Posovszky, P. THP-1 macrophages and SGBS adipocytes – a new human in vitro model system of inflamed adipose tissue. Front. Endocrinol. 2, 89 (2011).
Hotamisligil, G. S. & Bernlohr, D. A. Metabolic functions of FABPs-mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 11, 592–605 (2015).
Koppen, A. & Kalkhoven, E. Brown vs white adipocytes: the PPARgamma coregulator story. FEBS Lett. 584, 3250–3259 (2010).
Lehrke, M. & Lazar, M. A. The many faces of PPARgamma. Cell 123, 993–999 (2005).
Schoonjans, K. et al. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 15, 5336–5348 (1996).
Matsuki, T., Horai, R., Sudo, K. & Iwakura, Y. IL-1 Plays an important role in lipid metabolism by regulating insulin levels under physiological conditions. J. Exp. Med. 198, 877–888 (2003).
Glund, S. & Krook, A. Role of interleukin-6 signalling in glucose and lipid metabolism. Acta Physiol. (Oxf.) 192, 37–48 (2008).
Jovinge, S. et al. Evidence for a role of tumor necrosis factor alpha in disturbances of triglyceride and glucose metabolism predisposing to coronary heart disease. Metabolism 47, 113–118 (1998).
Nov, O. et al. Interleukin-1β regulates fat-liver crosstalk in obesity by auto-paracrine modulation of adipose tissue inflammation and expandability. PLoS One 8, e53626 (2013).
Kawakami, M. et al. Human recombinant TNF suppresses lipoprotein lipase activity and stimulates lipolysis in 3T3-L1 cells. J. Biochem. 101, 331–338 (1987).
Hardardottir, I., Moser, A. H., Memon, R., Grunfeld, C. & Feingold, K. R. Effects of TNF, IL-1, and the combination of both cytokines on cholesterol metabolism in Syrian hamsters. Lymphokine Cytokine Res. 13, 161–166 (1994).
Greenberg, A. S. et al. Interleukin 6 reduces lipoprotein lipase activity in adipose tissue of mice in vivo and in 3T3-L1 adipocytes: a possible role for interleukin 6 in cancer cachexia. Cancer Res. 52, 4113–4116 (1992).
Hammond, M. E. et al. IL-8 induces neutrophil chemotaxis predominantly via type I IL-8 receptors. J. Immunol. 155, 1428–1433 (1995).
Turner, M. D., Nedjai, B., Hurst, T. & Pennington, D. J. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 1843, 2563–2582 (2014).
Bonecchi, R. et al. Induction of functional IL-8 receptors by IL-4 and IL-13 in human monocytes. J. Immunol. 164, 3862–3869 (2000).
Preobrazhensky, A. A. et al. Monocyte chemotactic protein-1 receptor CCR2B is a glycoprotein that has tyrosine sulfation in a conserved extracellular N-terminal region. J. Immunol. 165, 5295–5303 (2000).
Blazek, K. et al. IFN-lambda resolves inflammation via suppression of neutrophil infiltration and IL-1beta production. J. Exp. Med. 212, 845–853 (2015).
Chrysanthopoulou, A. et al. Interferon lambda1/IL-29 and inorganic polyphosphate are novel regulators of neutrophil-driven thromboinflammation. J. Pathol. 243, 111–122 (2017).
Juge-Aubry, C. E. et al. Adipose tissue is a regulated source of interleukin-10. Cytokine 29, 270–274 (2005).
Walter, M. R. The molecular basis of IL-10 function: from receptor structure to the onset of signaling. Curr. Top. Microbiol. Immunol. 380, 191–212 (2014).
Liu, B. S., Janssen, H. L. & Boonstra, A. Type I and III interferons enhance IL-10R expression on human monocytes and macrophages, resulting in IL-10-mediated suppression of TLR-induced IL-12. Eur. J. Immunol. 42, 2431–2440 (2012).
Liu, B. S., Janssen, H. L. & Boonstra, A. IL-29 and IFNalpha differ in their ability to modulate IL-12 production by TLR-activated human macrophages and exhibit differential regulation of the IFNgamma receptor expression. Blood 117, 2385–2395 (2011).
Strissel, K. J. et al. T-cell recruitment and Th1 polarization in adipose tissue during diet-induced obesity in C57BL/6 mice. Obesity 18, 1918–1925 (2010).
Dai, J., Megjugorac, N. J., Gallagher, G. E., Yu, R. Y. & Gallagher, G. IFN-lambda1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood 113, 5829–5838 (2009).
Singh, S. et al. Obesity and response to anti-tumor necrosis factor-α agents in patients with select immune-mediated inflammatory diseases: a systematic review and meta-analysis. PLoS One 13, e0195123 (2018).
Ofei, F., Hurel, S., Newkirk, J., Sopwith, M. & Taylor, R. Effects of an engineered human anti–TNF-α antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45, 881–885 (1996).
Aharon-Hananel, G., Jörns, A., Lenzen, S., Raz, I. & Weksler-Zangen, S. Antidiabetic effect of interleukin-1β antibody therapy through β-cell protection in the Cohen diabetes-sensitive rat. Diabetes 64, 1780–1785 (2015).
Owyang, A. M. et al. XOMA 052, an anti-IL-1β monoclonal antibody, improves glucose control and β-cell function in the diet-induced obesity mouse model. Endocrinology 151, 2515–2527 (2010).
Hagberg, C. E. et al. Flow cytometry of mouse and human adipocytes for the analysis of browning and cellular heterogeneity. Cell Rep. 24, 2746–2756 (2018).
Acknowledgements
We are grateful to Professor Martin Wabitsch (University of Ulm, Germany) and Professor Peter Staeheli (Medical Center University of Freiburg, Germany) for kindly providing SGBS preadipocyte cell line and IL-28R1−/− mice, respectively. This work was supported by the Ministry of Science and Technology of Taiwan (MOST-106-2311-B-006-008-MY2 and MOST-108-2320-B-006-052).
Author information
Authors and Affiliations
Contributions
Y.-H.H conceived and supervised the study. Y.-H.H, T.-Y.L and C.-J.C designed the experiments, interpreted the results, and generated the figures. C.-H.K and C.-H.W collected the clinical samples and analyzed the data. Y.-H.H and T.-Y.L wrote the manuscript. Y.-H.H, T.-Y.L, C.-J.C, F.-H.C, and Y.-C.S performed the experiments and analyzed the data. All authors discussed the data and commented on the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, TY., Chiu, CJ., Kuan, CH. et al. IL-29 promoted obesity-induced inflammation and insulin resistance. Cell Mol Immunol 17, 369–379 (2020). https://doi.org/10.1038/s41423-019-0262-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-019-0262-9
Keywords
This article is cited by
-
IL-33 regulates adipogenesis via Wnt/β-catenin/PPAR-γ signaling pathway in preadipocytes
Journal of Translational Medicine (2024)
-
New treatment alternatives for primary and metastatic colorectal cancer by an integrated transcriptome and network analyses
Scientific Reports (2024)
-
Low-grade inflammation is associated with a heterogeneous lipoprotein subclass profile in an apparently healthy population sample
Lipids in Health and Disease (2023)
-
Interleukin-29 level in psoriasis before and after narrow-band ultraviolet B and its relationship with metabolic syndrome
Archives of Dermatological Research (2023)
-
20 Years with SGBS cells - a versatile in vitro model of human adipocyte biology
International Journal of Obesity (2022)
